
What Are Virtual Cells?
Learning "universal representations" of life's fundamental unit

Welcome to The Century of Biology! This newsletter explores data, companies, and ideas from the frontier of biology. You can subscribe for free to have the next post delivered to your inbox:
Before jumping into today’s essay, I’ve got an update to share.
I’ve joined Amplify Partners to help build a $200M early-stage venture capital fund dedicated to technical founders in bio. It’s pretty obvious that this has been my obsession for years—I’ve got receipts! We are building a firm in service of the next generation of technical founders like Neil Kumar at BridgeBio, Eric Kelsic at Dyno Therapeutics, Simon Burns at Vial, Viswa Colluru at Enveda, and more. There is so much left to build…
For more, you can read our Manifesto or check out the news coverage.
This is also a good reminder: I’m not a journalist! My goal is not to record events from a detached and objective vantage point—like the incredible cameras that hover over the action in modern sports stadiums. I’ve got opinions. Even worse, I’ve got positions in companies, and relationships with many of the people in my essays.
In other words, I’m inextricably linked to the subject—accelerating biotech is my life’s work, after all. Just as before, I’ll specifically highlight when I’m writing about companies that we’ve decided to partner with.
Lastly, I’ve got a message for Packy McCormick: thank you, thank you, thank you. I learned an extraordinary amount working at Not Boring. About writing and investing, sure. But also, life. Packy is one of the smartest and most generous people I’ve ever met. Despite being a New Yorker, Packy is the clearest embodiment of Silicon Valley positive sum thinking. It feels like he is hardwired to use his platform and gifts as a writer to boost others. I describe Packy as “chaotic good.” He’s a prolific writer, absurdly fast marathoner, brother, husband, dad, and friend. I’m not sure where he finds the energy to do it all, but I know that I’ll be a lifelong reader and Packy fan. Thank you.
Alright, let’s jump in! 🧬

On August 4th, 2017, Andrej Karpathy told us something profound about where the world was going in a cryptic tweet: “Gradient descent can write code better than you. I’m sorry.”
He was talking about the magic of deep learning—a subject he knew a lot about. At the time, Karpathy was leading Tesla’s autopilot efforts. By then, he’d spent over a decade obsessed with this subfield of AI, beginning his exploration well before its string of sweeping breakthroughs and Nobel Prizes. When he started out, the field was a backwater academic discipline.
As an undergrad at the University of Toronto, he studied under Geoff Hinton—now considered “the godfather of AI”—and attended his small reading groups on the subject. I can only imagine Hinton, a tireless, rail thin professor who claims to have literally never sat down since 2005, proselytizing to a young Karpathy in a practically empty university class room about a secret that few people yet understood.
The secret was that artificial neural networks—especially deep neural networks—are algorithms that can learn complex computer programs from data.
Consider an image classifier. How would you implement a program to determine if an image contained a kitten? Maybe you’d start by looking for patterns of pixels that looked like the outline of a face with pointy ears. But this would be brittle and would require thousands of lines of code accounting for different orientations and edge cases. Extending beyond kittens would quickly get out of hand.
Instead of writing code for an image classifier, we can train neural networks to do this by providing pairs of images and their corresponding labels. With sufficient data, the learned classifiers can even surpass human performance on the same task. The deep kernel of truth in Karpathy’s tweet was that gradient descent—the optimization algorithm responsible for tuning the networks to learn this type of mapping—could produce programs that no human could replicate.
In a subsequent blog post entitled Software 2.0, he produced a simple diagram describing this concept that has been etched into my own neural network ever since.

Software 1.0 consists of the set of programs—like terminals, word processors, or Web browsers—that humans can write by hand. Software 2.0 is the term Karpathy uses to describe the expanded set of programs that can be learned by neural networks with optimization algorithms (such as gradient descent).
Karpathy continued, “It turns out that a large portion of real-world problems have the property that it is significantly easier to collect the data (or more generally, identify a desirable behavior) than to explicitly write the program.”
Because of this, deep learning—which is now basically synonymous with AI—has steadily marched across many disciplines of computer science delivering world-class results. Even for problems that seem very much like traditional software 1.0, such as sorting and hashing, algorithms are now being rewritten by AI.
And of course, AI can deliver product experiences that simply aren’t possible to implement with traditional engineering. ChatGPT lives very far outside of the program space accessible via human coding. This paradigm shift has already generated trillions of dollars of market value.
Karpathy was right.
Gradient descent can write code better than you. I’m sorry.
Few disciplines feel this more acutely than biology.
A longstanding joke about biology has been that if you enjoy science but are bad at math, it’s a great major. Molecular biology is not like physics. The complexity—and nonlinearity—of living systems has required a brute force approach to knowledge creation and dissemination. Biology textbooks are full of facts and cartoons, not equations.
AI is starting to change this in a big way. After decades of research—including lots of attempted math and software 1.0—the problem of 3D protein structure prediction was effectively solved by deep learning.1 As Karpathy said, many real-world problems are easier to solve by collecting data rather than attempting to write programs. This is definitely one of those problems.

This breakthrough—and the resulting progress on the inverse problem of protein design—earned David Baker, Demis Hassabis, and John Jumper the 2024 Nobel Prize in Chemistry.
It’s not too hard to understand the logic behind this. Few macromolecules are as important as proteins. Delivering a step change in our ability to resolve and design their structures is a big deal.
With all the excitement and progress, protein modeling is now one of the fastest moving areas of biological research. As Brian Naughton has chronicled, our ability to design protein binders is rapidly improving. One way to think about this is that the binding interaction between two molecules—either proteins or ligands—is a logical next step for molecular machine learning.
But what about entire complexes of molecules, or even interactions between those complexes?
In 2023, I speculated about this, saying, “We already have powerful models of specific parts. Now we’re learning models of parts composed together into circuits. What about models of combinations of circuits—getting us closer to predictive models of whole cells? Or combinations of cells—getting us closer to models of tissues? Can we compose the outputs of our BioML models together like a set of Matryoshka dolls?”
Even broken clocks are right twice a day. This does seem to be where research efforts are now headed. There has been a palpable uptick in focus on a longstanding Holy Grail of biology: effectively simulating the behavior of cells.
Top labs and companies around the world are now racing to build the “Virtual Cell,” a concept that has been around since the turn of the century. With new tools, the big bet is that this vision is now within reach.
This concept is like an onion. At the highest level, “virtual cell” is a catch-all term for garnering excitement—and funding—that is abstract enough to mean many things to many different people. Basically, a buzz word.
Going one layer deeper, researchers are producing perspective articles—the scientific equivalent of manifestos—laying out the shape of more specific research programs. A particularly important example is the recent paper How to build the virtual cell with artificial intelligence: Priorities and opportunities, which we’ll soon explore in more detail.
At the core of the onion, of course, are the actual research results. We’re seeing the first wave of architecture innovation—and importantly, benchmarking—and several concerted efforts to scale data generation.
But before we start peeling back the onion on AI virtual cells, let’s consider the alternative approach. What it might look like to directly write down math to describe cells?
“A Leap of Faith”
Coming of age in the Bay Area in the 1980s, Markus Covert remembers a special blend of anarchy and excellence all around him. There were few rules and none of the extreme pressures around crafting the perfect set of “extracurriculars” that kids face today. But intellectual stimulation was easy to find. He’d end a day of aimless wandering by stopping over to visit a friend whose family who had just bought the first Macintosh. The sleepy suburbs of Silicon Valley were evolving into the tech mecca of the world.
In university, Covert decided to study chemical engineering, partially because of his longstanding interest in science, partially because it was considered to be the hardest major on campus. It lived up to its reputation: it was really hard. But the payoff wasn’t quite worth it. Modeling petrochemical plants wasn’t particularly awe-inspiring.
Underwhelmed by chemical engineering, Covert searched for the small set of graduate programs in biological engineering that were offered at the time. He found his way to U.C. San Diego, which had launched the first bioengineering department in the entire U.C. system just a few years earlier in 1994. This proved to be a wise choice.
At the time, the field of genomics was just starting to coalesce. One of the centers of excellence—and plenty of anarchy—was San Diego. Craig Venter had recently established The Institute for Genome Research, a private research center that was tackling seemingly insane projects.
Venter, an extraordinarily forward-thinking and competitive scientist who would later become famous for racing against the public Human Genome Project, assembled a small group of scientists who matched his intensity and ambition.
In successive landmark papers, they produced the first genomic map of a free-living organism, the genome of the world’s smallest organism (Mycoplasma genitalium), and the genome of a famous gut pathogen.2

But the vision extended far beyond genome sequencing. Venter and his team pushed forward a research agenda to simulate entire cells on a computer and ultimately even synthesize their genomes. Sequencing. Synthesis. Simulation. These ideas hung together as part of a mission to engineer life in a way that was fundamentally new.
Covert remembers this work blowing his mind. “To me and other young biologists in the late 1990s, this gang was Led Zeppelin: iconoclastic, larger-than-life personalities playing music we had never heard before.”
In his own research, Covert worked under Bernhard Palsson, one of the early pioneers of computer modeling in biology. It was a perfect intellectual fit. He became obsessed with the beauty of generating verifiable predictions. For his main project, Covert produced an important paper connecting Palsson’s techniques for modeling cellular metabolism with approaches for representing gene regulation.
After an initial academic faculty search with little success, Covert convinced David Baltimore—another giant of modern biology—to take him on as a postdoc. After earning the Nobel Prize at age 37 for his work on reverse transcription—which required revising the Central Dogma of molecular biology—Baltimore became a scientific leader. He helped establish the Whitehead Institute before serving as the president of Rockefeller University and, ultimately, the president of Caltech.
Initially, Baltimore was skeptical of Covert’s Venter-inspired “systems biology” ideas. Covert remembers chasing after Baltimore—who was whipping a Segway prototype around the Caltech campus—as he advocated for model-driven biological research. Finally, Baltimore relented, saying “We’ve never had somebody with your type of background in the lab before, so maybe that’s reason enough.”
At Caltech, Covert fell in love with experimentation. He learned how to combine his modeling techniques with meticulous experiments tracking individual transcription factors in actual cells. Crucially, he was exposed to the early prototypes for live-cell imaging technology. In order to learn, Covert volunteered to monitor the microscopes overnight, holding together a makeshift rig consisting of a chicken egg incubator, cardboard, and duct tape, making sure the temperature remained a consistent 37 degrees to keep the cells alive and happy.
Finally, Covert got the chance to move back to the Bay to start his own lab at Stanford. He continued his work on computer modeling and live-cell imaging. But there was a deeper question below the surface.
What would it take to simulate a cell?
In 1984, a Yale biophysicist named Harold Morowitz had laid out a plan of attack in an article entitled The completeness of molecular biology.
First, sequence the smallest organism, a Mycosplasma bacterium. Covert’s former neighbors in San Diego at the Venter Institute had already done this. Check.
Next, simulate the behavior of each of the ~600 genes estimated to be in the genome. Morowitz reasoned, “At 600 steps, a computer model is feasible, and every experiment that can be carried out in the laboratory can also be carried out on the computer. The extent to which these match measures the completeness of the paradigm of molecular biology.”
In practice, this step proved to be much harder than expected. Venter and Clyde Hutchinson’s early prototypes represented only 127 genes in the M. genitalium genome and the simulation results barely resembled experiments. The paradigm of molecular biology felt far from complete.
Biking home on Valentine’s Day in 2008, Covert mulled this over. Suddenly, discrete ideas collected throughout all of his training started clicking together.
At Caltech, he had stared down the microscope at individual cells. A whole-cell simulation should aim to approximate one cell rather than a population of cells. And similar to his thesis work, it would require integrating distinct mathematical representations for different cellular processes into one model. Thinking back to his chemical engineering days, he thought of HYSYS, a chemical process simulator composed of discrete modules.
He immediately got to work sketching out modules for each cellular process in a single M. genitalium cell. Things started to snowball. Covert recruited two graduate students, Jonathan Karr and Jayodita Sanghvi, who were ambitious (and maybe, crazy) enough to sign up for this new undertaking.
This was not exactly glamorous work. Karr, Sanghvi, and Covert spent two years visiting multiple research libraries to physically scan nearly a thousand research papers. They were looking for any crumb of molecular information about M. genitalium they could get their hands on. All of these data points, along with theoretical assumptions and measurements from other species, were stored in a MySQL database.
As they expanded and refined their modeling efforts, the team grew. Covert recruited researchers with a variety of different backgrounds. One crucial addition was Jared Jacobs, a childhood friend of Covert who had gone on to become a talented software engineer at Google. Before joining a new company, Jacobs took a leave of absence to help out for a few months.3
With this influence, the team adopted test-driven development and moved towards an object-oriented programming model. Each discrete cellular process and molecule was represented as its own object to help grapple with the complexity of the problem.4
Another essential modeling assumption was that “even though all these biological processes occur simultaneously in a living cell, their actions are effectively independent over periods of less than a second.” This meant that each module—of which there were 28 in total—could be executed independently for each one second interval. Information that needed to flow between modules could be exchanged between these discrete time steps forward.
Years into this project, there was no empirical evidence that this crucial assumption—or many others that were baked into the model—would actually yield results. As Covert recalls, “it was a leap of faith.”
It’s the type of modeling problem that “would drive mathematicians insane,” as Covert puts it. There is no bounded solution. “But that’s where engineers shine.”
The model started out with abysmal performance. Simulations had little to no correspondence with reality. But after over a year of hacking, tinkering, and tuning, experimental and digital results started to converge. It was gradient descent by hand.
The final product, published in Cell in 2012—four years after Covert’s initial spark of inspiration—was extraordinary.5 Every single annotated gene was accounted for. Distinct mathematical tools were matched with each cellular process.6

While far from perfect, the whole-cell model approximated cell growth and division, produced values within an order of magnitude of a wide range of metabolic data, and correlated with experimental gene expression data.
It was a big step forward. A central design element was the incorporation of “sensors” that read out cell state at each step of the simulation. With this digital measurement apparatus, they explored a wide range of biological questions with their model.
This was true “model-driven biological discovery.” As Horowitz had envisioned, each discrepancy between prediction and experiment was a chance to move closer to a “complete” understanding of molecular biology.
And there were many discrepancies. The authors wholly acknowledged this, saying, “similar to the first reports of the human genome sequence, the model presented here is a ‘first draft,’ and extensive effort is required before the model can be considered complete.”
One major bottleneck in making further progress was the lack of experimental tractability of M. genitalium compared to other model organisms. The organism’s small genome helped reduce the scope of the challenge for the first whole-cell model. But M. genitalium’s small cellular size, penchant for antibiotic resistance, and lack of established tools made it impossible to validate certain predictions in the lab.
In the thirteen years since this work, Covert’s lab and others have marched forward on the problem of modeling E. coli. While E. coli is one of the simplest model organisms, its genome is nearly an order of magnitude larger than M. genitalium (4,641,652 versus 580,070 base pairs) and contains over 4,000 genes. In 2021, the E. coli whole-cell model represented 43% of these genes.
Covert thinks considerable progress has been made since then. He believes we are approaching a “Turing test moment” where no E. coli biologist could reliably detect the difference between the outputs of the simulation and a matched experiment.
For one of the most exhaustively studied microbes, simulation and reality may soon be indistinguishable.
For human cells, with billions of DNA base pairs and tens of thousands of genes, the timeline for mechanistic whole-cell models is much less certain. It may still be a leap of faith that it’s even possible.
Which brings us back to gradient descent. What if computer models of cells are the quintessential type of program that we should be learning from data rather than attempting to define and parameterize by hand?
The Bitter Lesson
Silicon Valley has changed since the 1980s. While the built environment has remained frustratingly consistent, tech has exploded. Of the top ten largest companies in the world, seven are now technology companies. A new cultural intensity exists—for technologists and for their kids—that Covert doesn’t remember being present growing up. Maybe the ratio of excellence and anarchy has shifted. Or maybe the expectations are heightened.
With great power comes great responsibility.
In the 20th century, legendary American philanthropists such as Andrew Carnegie, John D. Rockefeller, and Henry Ford contributed unprecedented sums of money to public works projects. Entirely new universities, research institutions, libraries, and hospitals were established. According to the historian Olivier Zunz, “The philanthropic projects were acts of generosity and hubris on a scale never before entertained.”
These are big shoes to fill for the new Tech Elite, who are now grappling with how to best exercise their newfound power. Much like their predecessors, an early focus of Silicon Valley philanthropists has been scientific research. History doesn’t repeat, but it often rhymes.
A central example is the Chan Zuckerburg Initiative. Founded in 2015, Mark Zuckerberg and Priscilla Chan announced they would be donating 99% of their Facebook (now Meta) shares via the CZI’s efforts.
The CZI’s first major project was the establishment of a $3B Biohub that would provide new resources and connective tissue between the Bay Area’s three major biomedical research institutions: UCSF, Berkeley, and Stanford.7
The Biohub’s founding mission is to “cure, prevent, and manage all diseases by the end of this century,” which was immediately polarizing.8 Zunz could have just as easily been writing about this project.
At first, scientists—including Steve Quake, the CZI’s first Head of Science—laughed at the idea. Quake joked, “I couldn’t say it with a straight face … I don’t know why you hired me, because I couldn’t say the mission.”
But gradually, the idea grew on him and other scientists. With most research relying on a bureaucratic and increasingly conservative NIH, a new institution was a good thing. And crucially, a longer time horizon for funding meant that scientists could take bolder bets. Stacking these types of projects together over a century, who knows what could happen?
Specifically, the CZI aims to think on ten year time horizons—over three times longer than the average NIH grant. But a decade isn’t long enough to completely omit deliverables and clear milestones.
According to Quake, the CZI’s “North Star for the next decade is understanding the mysteries of the cell.”
The rationale was twofold.
First, Quake and others at the CZI had seen the march of gradient descent that Karpathy so succinctly described. AI is happening. They felt that proteins were the first breakthrough application of these techniques in biology, but they wouldn’t be the last.
Second, AI models need a lot of data. AlphaFold and subsequent protein models were only possible because of the data bank of crystal structures that researchers had contributed to for decades. When it came to cellular data, the CZI had been investing heavily in the development of enormous atlases of single-cell genomics measurements.
Maybe these massive single-cell datasets could fuel a step change in the performance of AI models for predicting cell behavior.
In March of 2024, CZI’s AI team, led by Theofanis Karaletsos, got to work. The first order of business was to host a workshop at the San Francisco Biohub to start sketching out this thesis in more detail. They convened a group of leading AI researchers and single-cell biologists to consider what might be possible.

Ultimately, conversations from the workshop helped to unify ideas that were being developed in each lab. Over the course of several months, these ideas were distilled into a perspective in Cell entitled How to build the virtual cell with artificial intelligence: Priorities and opportunities.
In the introduction, the authors outline prior efforts in cellular simulation. Covert’s “pioneering work” modeling M. genitalium in 2012 is cited as a major milestone. But the roadblocks for bottoms-up mechanistic approaches are enumerated.
Cells are composed of a diverse set of exquisitely complex processes. Each process operates across scales ranging from atoms to entire tissue systems in the body. To make matters even worse, cellular behavior is often nonlinear, meaning subtle differences in signaling can drive massive changes downstream.
Can we ever hope to produce a complete bottoms-up mathematical description of mammalian cells? When?
Instead, the authors propose a different approach: “Two exciting revolutions in science and technology—in AI and in omics—now enable the construction of cell models learned directly from data.”
If they are right, it wouldn’t be the first time. In practically every domain of computer modeling with sufficient data, learned models have outperformed more detailed mechanistic models. It’s what Rich Sutton, a Canadian AI researcher, calls “the Bitter Lesson.”
In a 2019 blog post that’s become famous in the AI community, Sutton wrote:
The biggest lesson that can be read from 70 years of AI research is that general methods that leverage computation are ultimately the most effective, and by a large margin. The ultimate reason for this is Moore's law, or rather its generalization of continued exponentially falling cost per unit of computation. Most AI research has been conducted as if the computation available to the agent were constant (in which case leveraging human knowledge would be one of the only ways to improve performance) but, over a slightly longer time than a typical research project, massively more computation inevitably becomes available. Seeking an improvement that makes a difference in the shorter term, researchers seek to leverage their human knowledge of the domain, but the only thing that matters in the long run is the leveraging of computation. These two need not run counter to each other, but in practice they tend to. Time spent on one is time not spent on the other. There are psychological commitments to investment in one approach or the other. And the human-knowledge approach tends to complicate methods in ways that make them less suited to taking advantage of general methods leveraging computation.
Consider natural language processing. Decades of research building exquisitely detailed models of linguistics and semantics produced models that were superseded by the Transformer. The Transformer is a general method that can scale with data and compute to learn rich representations of language.
Internet-scale data and compute won.
The argument sketched out in the CZI paper is that we should test the same recipe for cell biology. But instead of Internet data, we have omics to build on top of. And the improvements in sequencing technology have outpaced even Moore’s Law:

With the exponential improvements in DNA sequencing since the Human Genome Project, we can now produce atlases of single-cell measurements at an extraordinary scale. Rather than painstakingly developing mathematical models based on human knowledge, what if we attempted to learn cellular dynamics directly from this data?
In the proposed AI Virtual Cell (AIVC) approach, we should focus on using general learning methods to capture universal representations (URs) of each major cellular building block—such as DNA, RNA, and proteins.

Again, think about this as a big Matryoshka doll of different BioML models. A model for DNA. A model for RNA. A model for proteins. And a model to integrate this information into a snapshot of cellular behavior alongside data types like microscopy, proteomics, or RNA sequencing. And ultimately, connecting these models together in graphs to represent multi-cellular interactions.
The major research questions become:
What types of data do we need?
What are the right models for each data type?

Probably the most counterintuitive part of this strategy is what success would look like. Imagine that this works. We learn a general model that can accurately recapitulate cellular behavior.
We would have a model of cell biology—we just wouldn’t understand it!
To remedy this, the authors propose developing a suite of tools to perform computational experiments.
First, we would need manipulators. These tools would learn a representation of a perturbation—such as a chemical or genetic manipulation—that could be applied to a UR. A manipulator would convert one UR into another, representing a change in cell state.
But we would also need tools that produce human interpretable outputs, not just unintelligible vector representations of cell state. This is where decoders come in. These tools would learn to map between URs and synthetic experimental measurements, such as generated microscope images or sequencing data.

Essentially, we’d be swapping out physical cells for their virtual simulacra. The major benefit would be that virtual experiments scale in a way that physical experiments don’t. In theory, we could test billions or trillions of hypotheses using these models. If a result looks promising, it could be validated in physical reality.9
Like most perspectives, the AIVC paper attempts to unite many ideas that had already been swirling around. Many of the authors had spent years testing related approaches. Models like Universal Cell Embeddings, published in 2023, showed how protein models could be integrated with RNA data to create cellular representations. Other architectures like GEARS and scGPT were already developed to simulate cellular perturbations.
The central message was that this research direction—testing the Bitter Lesson for cell biology—is the most promising path forward. Driving this point home, the paper concludes by saying, “We believe that we are stepping into a new era of scientific exploration and understanding. The confluence of AI and biology, as encapsulated by AIVCs, signals a paradigm shift in biology and shines as a beacon of optimism for unraveling multiple mysteries of the cell.”
Natural Predators
Extraordinary claims require extraordinary evidence. Whenever a new modeling paradigm emerges, other scientists immediately attempt to poke holes in it. It’s like a form of immune response. And typically, the grander the claims, the faster—and larger—the response is.
As soon as AI cell models emerged, third party benchmarks and evaluations started to trickle out. Some of the results were quite surprising. In September of 2024, a group of highly experienced biostatisticians from Germany published a preprint comparing many of the first AI models for perturbation prediction against “deliberately simplistic” linear models.
Predicting the response to perturbations has become a major focus area for AIVC researchers. With experimental tools like CRISPR, specific genes can be up- or down-regulated, like turning a nob on a big cellular control panel. Using RNA sequencing, it’s possible to see what other genes were turned up or down based on that manipulation. Methods like Perturb-seq were invented to do this on a massive scale.
Basically, how does a cell switch up its gene expression programs when it is poked?
So the German lab evaluated how accurately the models could predict these responses for perturbations where two genes were up-regulated at the same time.
Two baseline models were used:
No change: in response to a double perturbation, this model would predict there would be no change to gene expression.
Additive model: simply return the sum of the change in expression for each single perturbation.10
Counterintuitively, the simple additive model had lower error rates than the complex AI models.

Separately, Eric Kernfeld produced similar results across a broader range of datasets during his thesis work at Johns Hopkins. In an accompanying blog post, Kernfeld wrote, “The natural predator of the methods developer is the benchmark developer, and a population boom of methods is naturally followed by a boom of benchmarks.”
The natural predators had found vulnerabilities.
AI models for these types of prediction tasks are clearly promising. The developers of the linear baseline wrote, “We do not consider our negative results on the foundation models’ performance in the prediction task as arguments against this line of research ... the progress brought by the transformer architecture and the transfer learning paradigm for many machine learning tasks is real and substantial.”
But the first implementations still felt far from producing “non-trivial insights,” as the benchmark developers put it.
I asked Yanay Rosen and Yusuf Roohani, two of the primary authors of the AIVC perspective, about how they interpreted these results. Both researchers wholeheartedly acknowledged the limitations of these early prototype models.
Rosen also pointed out that for the task of creating universal cell embeddings, AI models are already producing results that can’t be captured with simpler methods. These approaches make it possible to represent cells across diverse datasets, tissue types—and even species—in a single shared coordinate space. Biologists are already using these representations to discover new cell types and answer fundamental questions in evolutionary biology.
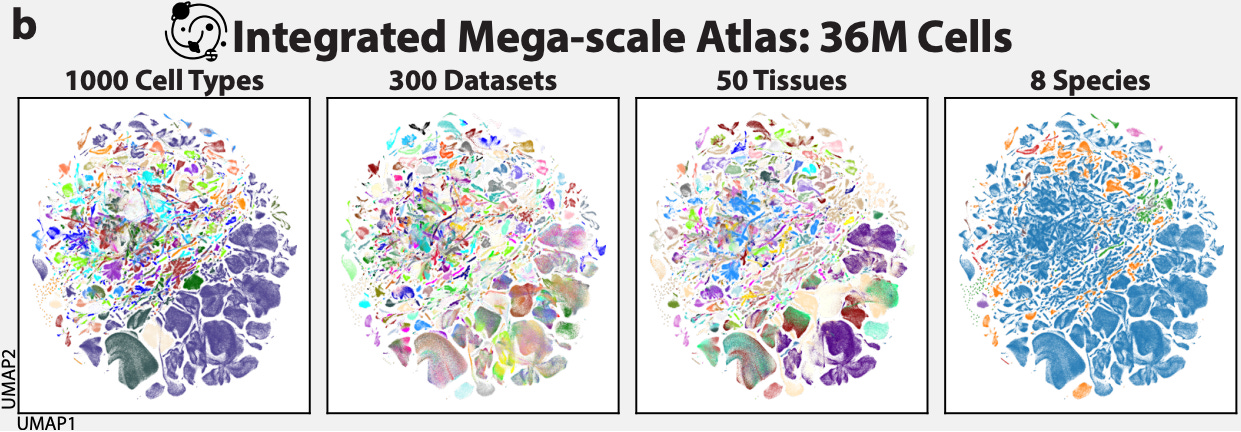
Recently, the AI team at the CZI extended this research direction with the release of the Transcriptformer model. Trained on over 112 million cells collected from 12 different species—representing 1.5 billion years of evolutionary distance—the model was designed for a wide range of prediction tasks. In their release, the CZI team wrote, “Researchers can use TranscriptFormer to predict what different types of cells are, whether a cell is diseased, and how genes interact.”
For the task of positioning a cell in relation to all other cells, there seems to be enough data for AI models to shine. But for predicting the dynamics of how a cell changes when perturbed, existing data and models appeared to be insufficient.
This is starting to change.
Roohani, who now leads a machine learning research group at the Arc Institute, is very excited about more closely integrating with experimentalists. The Arc, which is another product of large-scale tech philanthropy for science, has launched its own Virtual Cell Atlas project. A major focus is large-scale data generation.
This week, the Arc team published new results from this research program. Their new model, called State, appears to be a major step forward for perturbation prediction. You can see the interplay between benchmark developers and methods developer in action: linear baselines are now front and center. And this time around, the models appear to be picking up “non-trivial insights” more effectively.

{kind=link}
A key driver of the jump in performance comes from a more careful representation of the biological noise that is present in the data. Cells are fundamentally noisy, heterogenous systems—and the experiments interfacing with them bring their own limitations. The State model uses mathematical bookkeeping to account for the biological and experimental noise that can drown out true signal.
When they do that, something very important happens: the model’s performance appears to meaningfully increase with more data. In other words, Sutton’s Bitter Lesson starts to kick in.
Recently, Tahoe open-sourced a massive dataset measuring 60,000 drug perturbations in 100 million distinct cells—dwarfing all other publicly available single-cell datasets.11 After training on this scale of data and diversity of cellular contexts, the State model was able generalize to a wider range of perturbation predictions.
Signaling what we should expect over time, the authors wrote, “Overall, the performance and flexibility of State sets the stage for scaling the development of virtual cell models.”
So, what are virtual cells?
Let’s start with what they are not.
It’s hard not to hear this term and imagine a fancy graphical rendering of a cell in its gyrating three-dimensional glory. If you zoomed in, you’d see ribosomes meticulously examining RNA molecules as amino acid chains spooled outwards, or kinesin proteins marching along microtubule walkways.
Crucially, every piece of molecular machinery would behave the way that it would inside of an actual cell. It would be the Magic School Bus for grown-up cell biologists.
That is not what scientists are building—at least, not yet.
For Covert, the more appropriate analogy is a meteorological simulation. As he and his research team wrote in their own recent perspective, “numerical weather prediction is a comprehensive endeavor to integrate observations from around the world and in space, over multiple timescales, into a mathematical model which both holds an initial state of the global system and can produce forecasts of changes in the atmosphere several days in advance.”
He and others hope to produce an equivalent mathematical model capable of simulating microbial systems. And as Morowitz envisioned in the 1980s, every glitch in these models can be treated as negative space in our broader map of molecular biology.
Covert told me he “wants to find Neptune,” referring to the 19th century astronomer Urbain Le Verrier’s ability to discover an entirely new planet “with the point of his pen.”
AI researchers think about the problem differently. Guided by the Bitter Lesson, the goal is to specify as few priors as possible, letting the data speak. The first goal is prediction, not understanding.
The AIVC models are learning “the statistical shape of life,” as Karaletsos puts it. But this “shape” is imprinted in massive numerical vectors within these networks that are illegible to humans. In Geoff Hinton’s early deep learning courses, he would tell his students: “To deal with hyper-planes in a 14-dimensional space, visualize a 3-D space and say ‘fourteen’ to yourself very loudly. Everyone does it.”
What if these models achieve incredible predictive power? The real work would begin. Armed with virtual instruments, biologists would be able to manipulate cells at a scale that would be unimaginable in the lab.
If the Venter gang was like Zeppelin, these new research groups are more like Taylor Swift.12 Massive projects are produced back-to-back, accompanied by carefully coordinated press releases and social media announcements—which are essential for mimetic distribution in modern science.
Over time, it’s possible that these distinct sounds converge. The bottoms-up mechanistic modeling efforts may start to integrate AI methods into their systems. Covert’s lab was one of the first groups to develop deep learning methods for microscopy, after all.
Similarly, as AIVC research efforts expand beyond RNA, model architectures will become increasingly complex. Hierarchical systems with distinct modules for each biological process will start to resemble approaches from the world of whole-cell simulation.
For now, Roohani has a much simpler goal: make the existing models good enough that experimentalists adopt and use them. Like the “GPT Moment,” this may not require any semblance of perfection.
This could create a feedback loop of compounding progress as predictions are tested in labs around the world, creating more data to further refine the models.
Quake imagines a future where “cell biology goes from being 90% experimental and 10% computational to the other way around.”
The first step in that direction will be convincing researchers that they should at least run a query or two before picking up a pipette.
Notes and Further Reading
Much of this essay focused on whole-cell modeling efforts from the Covert Lab and early AIVC research from the authors of the Cell perspective:
Whole-cell modeling:
AI Virtual Cell:
How to build the virtual cell with artificial intelligence: Priorities and opportunities
Predicting transcriptional outcomes of novel multigene perturbations with GEARS
scGPT: toward building a foundation model for single-cell multi-omics using generative AI
Universal Cell Embeddings: A Foundation Model for Cell Biology
TranscriptFormer: A Generative Cross-Species Cell Atlas Across 1.5 Billion Years of Evolution
Predicting cellular responses to perturbation across diverse contexts with State
One of the tough things when writing about such an active space is avoiding the urge to be exhaustive. There is a lot more research happening in this space—including results that came out as I was writing:
Genentech has been pursuing this work for years under the direction of Aviv Regev, one of the AIVC authors:
Altos Labs has been working on perturbation prediction and producing benchmarks.
Recursion has consistently prioritized virtual cell research.
Last week, Xaira Therapeutics published a massive Perturb-seq dataset and announced their own efforts to build a virtual cell model.
Noetik has been building virtual cell models for spatial biology.
Theofanis Karaletsos also helped to lead research on perturbation prediction during his time at Insitro.
While seemingly less interested in the “virtual cell” terminology, NewLimit has been developing AI models to predict the effects of combinations of transcription factors (TFs). The goal is to identify TF cocktails that drive safe cellular rejuvenation.
This week, the Arc Institute announced a Virtual Cell Challenge.
Demis Hassabis has also prioritized virtual cell research as the logical next step for molecular machine learning. In a recent interview, he outlined a research plan focused on continually moving upwards from proteins to protein complexes, molecular pathways, and ultimately, cells. In some ways, this is a hybrid of the different ideas outlined in this essay!
This list is still far from exhaustive.
Thanks for reading this essay on the rapidly evolving world of virtual cells.
I’d like to thank Markus Covert, Yanay Rosen, Yusuf Roohani, and Theofanis Karaletsos for their helpful conversations as I was doing research.
If you don’t want to miss upcoming essays, you should consider subscribing for free to have them delivered to your inbox:
Until next time! 🧬
While the models perform extraordinarily well for a large portion of structure prediction tasks, they don’t quite behave as a universal solution yet. For example, most models still struggle to correctly predict the structural effect of variants in the protein sequence, or to understand the dynamics of the structure rather than a singular static snapshot.
The gut pathogen, Helicobacter pylori, was the pathogen that Barry Marshall and Robin Warren identified as the causal agent for gastric ulcers. To convince skeptics, Marshall drank a beaker of H. pylori, giving himself gastritis! Marshall and Warren were awarded the Nobel Prize in 2005 for their work.
For some engineers, a sabbatical or leave of absence means getting up to speed on one of the most challenging simulation efforts in the natural sciences. To each their own.
Alan Kay, one of the pioneers of OOP, got his original inspiration for the idea from biology. He later called Covert to talk about the whole-cell model. Many ideas in biology and computer science have this type of circular relationship—including AI!
I still distinctly remember coming across this paper for the first time in college and having my mind blown. If that’s not enough, Cell included it as one of the major landmark papers in the journal’s history, alongside the first reports of iPSC cells and CRISPR.
The supplement for this paper is insane. Each cellular process gets its own mathematical treatment. The metabolism module builds on Palsson’s flux balance analysis approaches. Events like DNA damage and the downstream repair mechanisms were modeled as stochastic processes and parameterized by kinetic data from experiments. I’m covering a sliver of the detail here, but it’s a rabbit hole worth going down.
I personally think this is awesome.
According to Priscilla Chan, it was Cori Bargmann who convinced them to even add the nuance of “prevent, and manage” to the mission statement.
I wrote about this shift towards learning and then reverse engineering models of biological function for DNA models back in 2021.
Specifically, it is “the sum of the individual log fold changes (LFC) compared to the control condition.”
We are very excited to have partnered with Tahoe at Amplify. More on this soon!
As a random aside, I couldn’t think of any major bands that have acclimated as successfully to the new world order of music distribution.
Deeply appreciate this article.
It all sounds so exciting. But, we have built a virtual WHALE that runs on a machine so old people reading this would fall over laughing. Yet, it works, with pure mathematics. And, along the way we learned about the huge gaps between what has been observed, and what we need to know to be able to create virtual organisms that survive.
It's entirely possible that the constraint of being severely underfunded forced us to be far more efficient in problem-solving and calls into question the funding model described here, as well as the approaches described. Their dependence on data is also their weakness because the knowledge is purely empirical (as "Metaclesus" points out in their remarks).