
Global importance analysis
A new neural network interpretation tool for genomics

Perhaps science does not develop by the accumulation of individual discoveries and inventions.
- Thomas S. Kuhn, The Structure of Scientific Revolutions
Overview
One of the ideas that has continually interested me is the paradigm shift. In 1962, the philosopher of science Thomas Kuhn published The Structure of Revolutions. This book introduced a powerful set of ideas about how progress in science happens.
Kuhn argued that scientific innovation is not a continuous march, but rather that it is punctuated by dramatic discontinuities. He identified four basic stages of knowledge generation in science:
1) Normal science: across a discipline, this is the stage in which there is a commonly agreed upon consensus about what constitutes a legitimate scientific explanation of a natural phenomenon. Given this consensus, researchers can march towards creating new explanations with homogenous approaches.
2) Extraordinary research: when groundbreaking discoveries are made, they can illuminate ways in which the current models in a field are limited. When discoveries evade being explained by the current concepts and techniques in a field, a discipline can enter a heightened stage of exploration and novel ideation.
3) Adoption of a new paradigm: over time, some of the new techniques, models, and tools proposed in the extraordinary research stage succeed in establishing a new paradigm that can successfully explain the new discoveries. This can be a brutal process. Max Planck explained this in dramatic fashion, saying “a new scientific truth does not triumph by convincing its opponents and making them see the light, but rather because its opponents eventually die, and a new generation grows up that is familiar with it.”
4) Aftermath of the scientific revolution: From a birds-eye view, these revolutions become compressed into a smooth narrative that is taught to the next generation of scientists in textbooks.
It is never easy to know in the current moment, but I would argue that we are in the middle of a stage of extraordinary research in genomics, and the life sciences more generally. In the post-genome era, we are challenged with the task of building new types of models to explain phenomena of enormous scale and complexity, such as the function and regulation of the human genome. Almost by definition, these problems require new approaches.
In thinking about how people approach building models, the statistician Leo Breiman wrote the article Statistical Modeling: The Two Cultures. In this paper, he argues that there are two primary camps when it comes to building models: “There are two cultures in the use of statistical modeling to reach conclusions from data. One assumes that the data are generated by a given stochastic data model. The other uses algorithmic models and treats the data mechanism as unknown.”
In the time since this was published in 2001, the deep learning revolution has swept many disciplines. Deep learning is exactly what Breiman calls algorithmic modeling. It is a modeling technique that relies on computational power and large amounts of data to build highly accurate predictive models, without needing to understand anything about the data generating process in question. This is often why deep learning models are referred to as “black box” models, where it is even unclear to the model developer how the model is generating predictions.
The predictive power of the models can be quite surprising. I distinctly remember a seminar where Erick Matsen described his astonishment at the predictive accuracy of a simple Keras model his graduate student created with a small amount of code on a daunting problem in phylogenetics. Joe Felsenstein, whose incredible work required carefully building hand-crafted models based on biology remarked “I don’t know whether to be excited or depressed.”
This represents one half of what I think might constitute a new scientific paradigm. These tools provide a powerful way to build highly accurate predictive models of systems without fully understanding them. Now, a discipline is emerging where a new generation of scientists is working on building the other half: tools to reverse engineer the models, to crack open the “black box” and actually use the models to drive discovery about the systems they can so accurately make predictions about.
One of the members of this new generation is Peter Koo, who is a new assistant professor at Cold Spring Harbor Laboratory. He has been working on developing new approaches to neural network interpretation for genomics. I am going to highlight the new paper from his group “Global importance analysis: An interpretability method to quantify importance of genomic features in deep neural networks” in the recent addition of PLOS Computational Biology.
Key Advances
It is worth starting out by thinking about what the entire workflow of this type of modeling looks like. In this paper, we are thinking about how to determine what sequences an RNA-binding protein (RBP) prefers.
We can divide the process into three stages:
Collect experimental measurements of which sequences RBPs preferentially bind.
Given the data, build a model that can accurately predict the likelihood of RBP binding for a specific sequence of RNA bases. For many years, the preferred technique was to leverage position-weight matrices (PWMs) or k-mer analysis1, but deep learning is now the state-of-the-art approach.
Use interpretability techniques to explore what the model has learned, and discover motifs2 and rules of RBP behavior.
We can see that this differs pretty considerably to other more hypothesis-driven workflows. There is no period of chin-scratching or pontificating before the data is collected. The process begins with data, and the rapid development of an effective predictive model using computation.
Once the model is built, it becomes the object of study and experimentation. Now the experiments are in silico. Experiments are run where the model input is systematically perturbed, such as in silico mutagenesis, where bases are digitally mutated to infer their impact on model prediction.
This paper introduces two primary innovations: 1) ResidualBind, a new convolutional neural network model for RBP prediction3, and 2) global importance analysis (GIA) a new interpretability method that “quantifies the population-level effect size that putative patterns have on model predictions.” We will briefly look at ResidualBind, and then focus on unpacking what GIA is and how it works.
Results
One thing that the field of machine learning does really well is formulate benchmarks. Competitions will be established with a specific dataset that is available to all researchers. For example, ImageNet is an image database that researchers continually used to compete to improve the predictive accuracy of computer vision algorithms. With a common goal for a field, the rate of progress can often be impressively steady.
For RNA-binding protein prediction, there is the RNAcompete dataset. Here is how ResidualBind performed:

I’m not going to dwell too much on the architecture or training of ResidualBind, other than saying that it is a convolutional neural network (CNN) that “takes one-hot encoded RNA sequence as input and outputs a single binding score prediction for an RBP.”4
The primary result of this paper is the introduction of the global importance analysis (GIA) interpretability method. Fundamentally, GIA is a formalized score that measures “the change in prediction caused by the presence of the pattern across a population of sequences.”5 Again, this is a tool for reverse engineering what sequence patterns are driving the model predictions. Calculating this association at the population level provides a much more durable interpretation, compared to predictions on individual sequences, which can vary.
What is really powerful about having such an effective predictive model is that you can swap it in as “a surrogate model for experimental measurements by generating data for synthetic sequences.” This is the key idea: after developing the GIA score for what constitutes a signal at the population level, “we opt to use a DNN, which has learned to approximate the underlying sequence-function relationship of the data, to “measure” the potential outcome of interventions (i.e. embedded patterns)—using predictions in lieu of experimental measurements.”
Now with a bit of intuition for GIA, we’ll look at one of the detailed analyses in the paper. The authors explored model predictions for the RBFOX1 protein, which has an experimentally verified motif of ‘UGCAUG’.
To interpret the predictions of ResidualBind, this study used in silico mutagenesis, which “systematically probes the effect size that each possible single nucleotide mutation in a given sequence has on model predictions.”

This shows two of the high scoring sequence logos from the mutagenesis. The first logo is an intact motif, while the other has two mismatches, indicating that the “number of motifs and possibly their spacing is relevant.”
Realistically though, “the sequence motif of an RBP is not known a priori, which makes the interpretation of in silico mutagenesis maps more challenging in practice.” Using an approach of embedding possible k-mers in sequences, the authors used GIA to discover the correct motif ab initio (from scratch):

With additional in silico experiments using GIA, we can also see that the model learns that having multiple binding sites is additive:
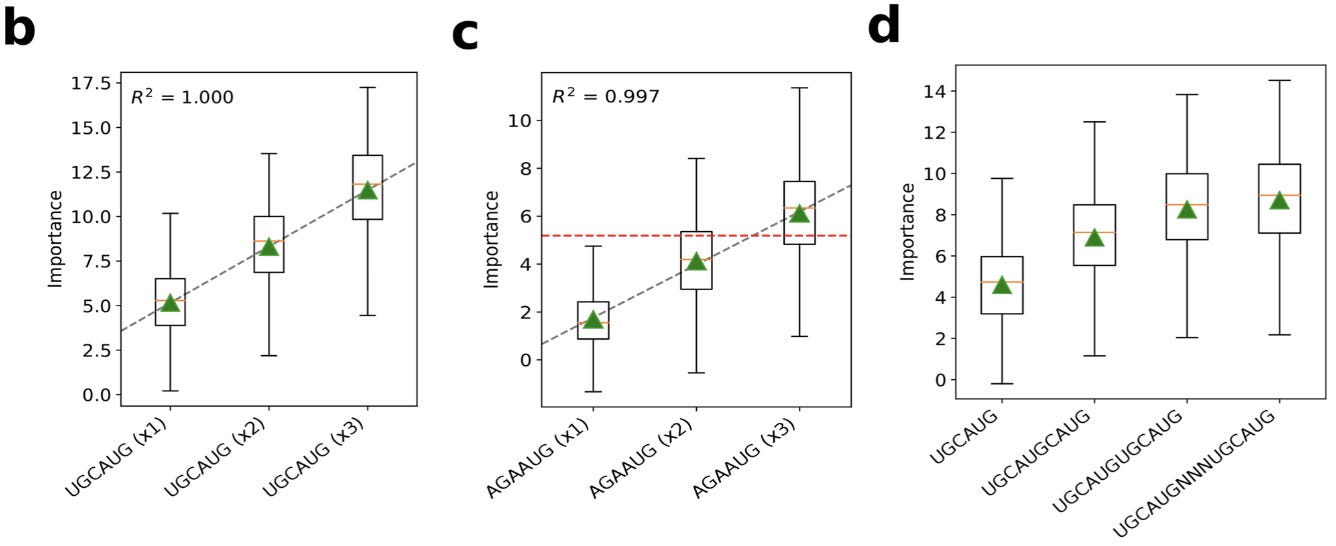
Here, the green triangles represent the global importance. This is interesting to think about in terms of what types of patterns these sequence models can capture.
As usual, there is a lot more interesting work to explore in this paper than I have highlighted here, and I would recommend interested readers to check out the other analyses.
Final Thoughts
In the science fiction novel Snow Crash by Neal Stephenson, there is a series of recurring scenes where the main character, Hiro Protagonist (yes, that is actually the character’s name), studies by having conversations with an AI librarian is his virtual reality office. Here is how this character is described:
The Librarian daemon looks like a pleasant, fiftyish, silver-haired, bearded man with bright blue eyes, wearing a V-neck sweater over a work shirt, with a coarsely woven, tweedy-looking wool tie. The tie is loosened, the sleeves pushed up. Even though he’s just a piece of software, he has a reason to be cheerful; he can move through the nearly infinite stacks of information in the Library with the agility of a spider dancing across a vast web of cross-references. The Librarian is the only piece of CIC software that costs even more than Earth; the only thing he can’t do is think.
This is an interesting depiction of AI. This type of system is an interesting artifact: like a vastly intelligent semantic search engine that can provide us with all of the answers, but none of the questions.
When present in the underlying biology, neural networks are able to capture rich non-additive interactions, the regulatory grammar and syntax of genomic sequences, and at ever-increasing distances, like in the work I highlighted recently. Now, tools such as GIA offer the promise of being able to begin to query these powerful AI models and decipher this information. It will be up to a new generation of scientists to wield these powerful tools and to ask the right questions.
Thanks for reading this highlight of “Global importance analysis: An interpretability method to quantify importance of genomic features in deep neural networks”. If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, have a great Sunday! 🧬
The k-mer is a substring of length k of a sequence. For example, ATC is a 3-mer of the sequence ATCG. This concept is widely used in bioinformatics.
A motif is a functional sequence. In this case, the motif is the set of bases that a RBP would recognize and bind.
This introduces an improvement in step 2 of the workflow above.
For folks with ML experience, this sentence should be pretty straightforward to parse. I’m not going to fully cover this here, but the gist is: the data consists of pairs of sequences and the RBP binding score. The sequence is encoded as a vector of 0s and 1s to for input into the model, which makes a prediction about whether or not the protein will bind to this sequence. This relationship is learned on training data, and then the performance is evaluated on test data, which are input pairs that the model hasn’t seen before.
If you want to dig deeper on the nuts and bolts of the score, the “Global Importance Analysis” section of the paper does a great job of presenting the two key equations that describe the score. I won’t unpack them here.