
Accelerating genetic design
Mixing and matching sequencing tools to build biological circuits faster

Welcome to The Century of Biology! This newsletter explores data, companies, and ideas from the frontier of biology. You can subscribe for free to have the next post delivered to your inbox:
If you enjoy this newsletter, it’s worth knowing that there is an entire conference dedicated to synthetic biology called SynBioBeta. You should consider joining, with the added benefit that I will be speaking this year! For a 10% discount on registration, you can use the code: H7TGP_JC10.
Enjoy! 🧬

The central theme of this newsletter is that multiple important technological paradigms are converging in the life sciences, and that it is a really big deal. The ramifications of this biotechnological progress will impact life on every scale. Our approaches to caring for our bodies and our planet could look very different by the end of the century. If we want to build a flourishing future, we should hope that they do.
Given the broad scope of this focus, the number of topics that I want to write about each week greatly exceeds the bandwidth that I have to write. Should I cover some of the incredible frontier science that I’m seeing, explore the evolution of biotech business models, analyze highly impactful companies, or dive into the histories of our most impactful biotechnologies? So far, I’ve been following my curiosity and working to grow as a writer by trying different approaches.
This week, I’m going back to the basics. I started this newsletter with a primary focus on writing about the most exciting new bioRxiv preprints that I read. Beyond explaining the science, my goal has been to give context for why I think it will be important. I haven’t done this in a while, but this week I came across a beautiful new study that really got me thinking. It is an excellent example of the deep integration between genomics and computation that is now possible.
The study that we’ll be exploring today is “Ultra-high throughput mapping of genetic design space” from the Bashor Lab at Rice University. This work knits several important threads together to introduce a new platform for rapidly testing genetic circuit behavior.
Alright, let’s jump in! 🧬
The goal: genetic circuits
I distinctly remember my first exposure to the concept of molecular circuits. I had just fallen down the biology rabbit hole, having changed university majors from English after being far more intrigued by the material.1 My life primarily consisted of spending time in the lab and reading new papers in order to get a sense for what was happening.
At a certain point, Aviv Regev came onto my radar. For the biotech uninitiated, Regev is one of the key pioneers of the highly impactful field of single-cell genomics, and currently leads R&D at Genentech. At the time, she was a scientist at the Broad Institute, where her lab focused on “biological circuits, gene regulation, and evolution.”
From the very inception of her career, cells were front and center. She trained at a unique and highly selective interdisciplinary program in Israel that let her direct her own studies. With this freedom, she chose to carve out a specialization in computational biology.
Specifically, Regev saw an enormous opportunity—while we had a quantitative framework for the transmission of genes and a fundamental model for how the macroscopic process of evolution works, we seemed to lack any robust and quantitative understanding of cells. This vision ultimately became her life’s work—as she developed the tools, datasets, and mathematical approaches necessary to describe the circuitry of cells.
Regev quickly became a major inspiration for me to learn computer science and become a computational biologist. As I explored this new world, I quickly realized that she wasn’t alone in her use of the language of circuits to describe how genes are composed together to accomplish complex cellular functions. A core focus of the entire field of synthetic biology is to be able to design new genetic circuits in order to be able to program cells to accomplish new goals.
It’s always helpful to consider examples. In synthetic biology, the lac operon from E. coli is pointed to as one of the first examples of a well characterized genetic circuit:

{kind=link}
This simple combination of genetic parts enables E. coli to switch up their metabolic machinery to consume lactose when they sense an absence of glucose. This type of genetic system that computes on inputs to change biological outputs is a genetic circuit. Generalizing from this, how can we systematically design genetic circuits for new tasks?

{kind=link}
How can we embed logic into genetic programs? An enormous amount of research has gone into this problem, and some truly impressive genetic circuits have been built. A stunning example of this is the design of a cell-scale biosynthetic pathway in baker’s yeast to produce scopolamine—a medicinally valuable chemical that acts as a neurotransmitter inhibitor.2
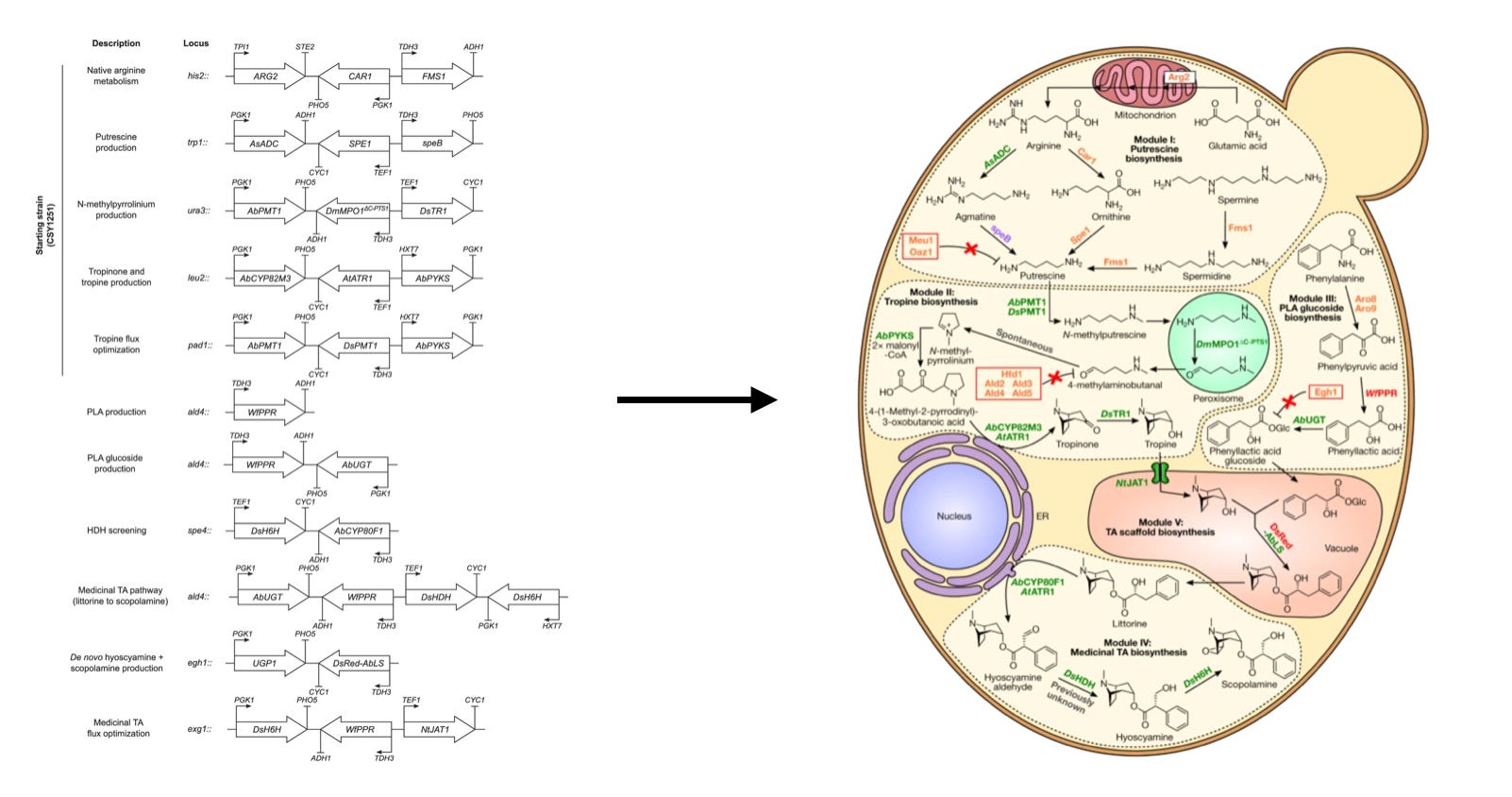
The sci-fi author William Gibson once said: “The future is already here. It’s just not evenly distributed yet.” This type of genetic engineering represents what we should be able to reliably to do in the future, but in practice the tools for doing this are still nascent. Many modeling frameworks and software tools have been developed for biological circuit design, but the sheer complexity of biology has limited their effectiveness.
This exact problem is what drove Aviv Regev to adopt the tools of genomics. She had started her career attempting to use a somewhat esoteric system called π-calculus from theoretical computer science to model cellular circuits. There was only one tangible result from this research direction—the insight that the lack of quality data was a massive bottleneck for effectively modeling complex cellular processes.
An article on Aviv Regev for MIT Tech Review put this really nicely:
“It’s like a complex computer that is made of these many, many different parts that are interacting with each other and telling each other what to do,” says Regev. The protein signaling networks are like “circuits”—and you can think about the cell “almost like a wiring diagram,” she says. But using computational approaches to understand their activity first requires gathering an enormous amount of data, which Regev has long done through RNA sequencing.
With large-scale gene expression data collected from millions of cells, computational modeling becomes a much different problem. Instead of using abstract calculus systems, it’s possible to start to learn the relationship between genetic circuits and the cellular phenotypes they are driving.
I think that this shift is long overdue for synthetic biology. A lot of thoughtful work has gone into developing frameworks for circuit design, but the experimental synthetic biologists building the most complex systems are still primarily relying on their hard-won empirical intuition. We need more data before the modeling work can truly begin. Have we put the cart before the horse?
How do we collect the data necessary to accurately model and design new genetic circuits?
Composition-to-function mappings
One of my main areas of focus is the 4-S model—where DNA Sequencing and Synthesis are being combined to produce large Scale data that requires new Software to wrangle and analyze it. The combination of all of these tools behaves like a new tech stack that is simultaneously accelerating many distinct avenues of biotechnological progress.
Some of the biggest wins for the 4-S approach so far have been for modeling sequence-to-structure relationships, or sequence-to-function relationships. The AlphaFold breakthrough is an example of using large-scale data and AI to learn the relationship between an amino acid sequence and its 3D structure. In synthetic biology, researchers have used genomics to develop datasets for many additional sequence-to-function relationships. We’ve seen this for promoters, terminators, transcription factors, RNA switches, and CAR-T signaling domains, to name a few examples.
All of these studies are examples of the fact that we’re getting a lot better at rationally designing molecules with all of these new tools. This type of work is moving from science to engineering, and a lot of exciting startups have been launched as the technology has improved.
Moving back to circuit terminology, each of these molecules will ultimately serve as parts within more complex programs. The scopolamine example mentioned earlier relied on protein engineering, but the protein was a part in a much larger system. So, how can we expand our 4-S stack to learn composition-to-function relationships between the distinct parts within a genetic circuit?
The platform introduced in this study attempts to solve this problem. It is a systematic process called CLASSIC that combines multiple types of sequencing technologies to characterize large numbers of genetic circuits:

Alright, so how does this work? The starting input is a large pool—a big group of distinct nucleic acids—of genetic parts, and a pool of barcode sequences. These inputs get stitched together using a very clever Golden Gate assembly protocol, and the resulting library of barcoded circuits gets sequenced using long-read nanopore sequencing. This step establishes a construct-to-barcode index. We now have molecular labels for each circuit.
Next, these circuits get tossed into mammalian cells and expressed. The phenotype of the circuits is measured using an approach called flow-seq. This works by first measuring the distribution of the phenotype, and then creating bins of cells within that distribution using flow cytometry. Each bin gets sequenced using short-read Illumina sequencing, which reads out the barcode. This creates a phenotype-to-barcode index.
Now, a given barcode is linked to two distinct pieces of information:
The composition of a genetic circuit
The phenotype of the genetic circuit
Linking these two pieces of information together is an example of going from composition to function. Now we’ve unpacked the different parts of the acronym that make this approach special—they are Combining Long- and Short- range Sequencing to Investigate genetic Complexity.
This system makes it possible to screen a massive number of different circuit combinations in a single pooled experiment. To demonstrate this system, they optimized a system called synTF which is a circuit that is under the control of a drug-inducible transcription factor:

Using CLASSIC, they profiled a circuit design space of 165,888 possible distinct configurations:
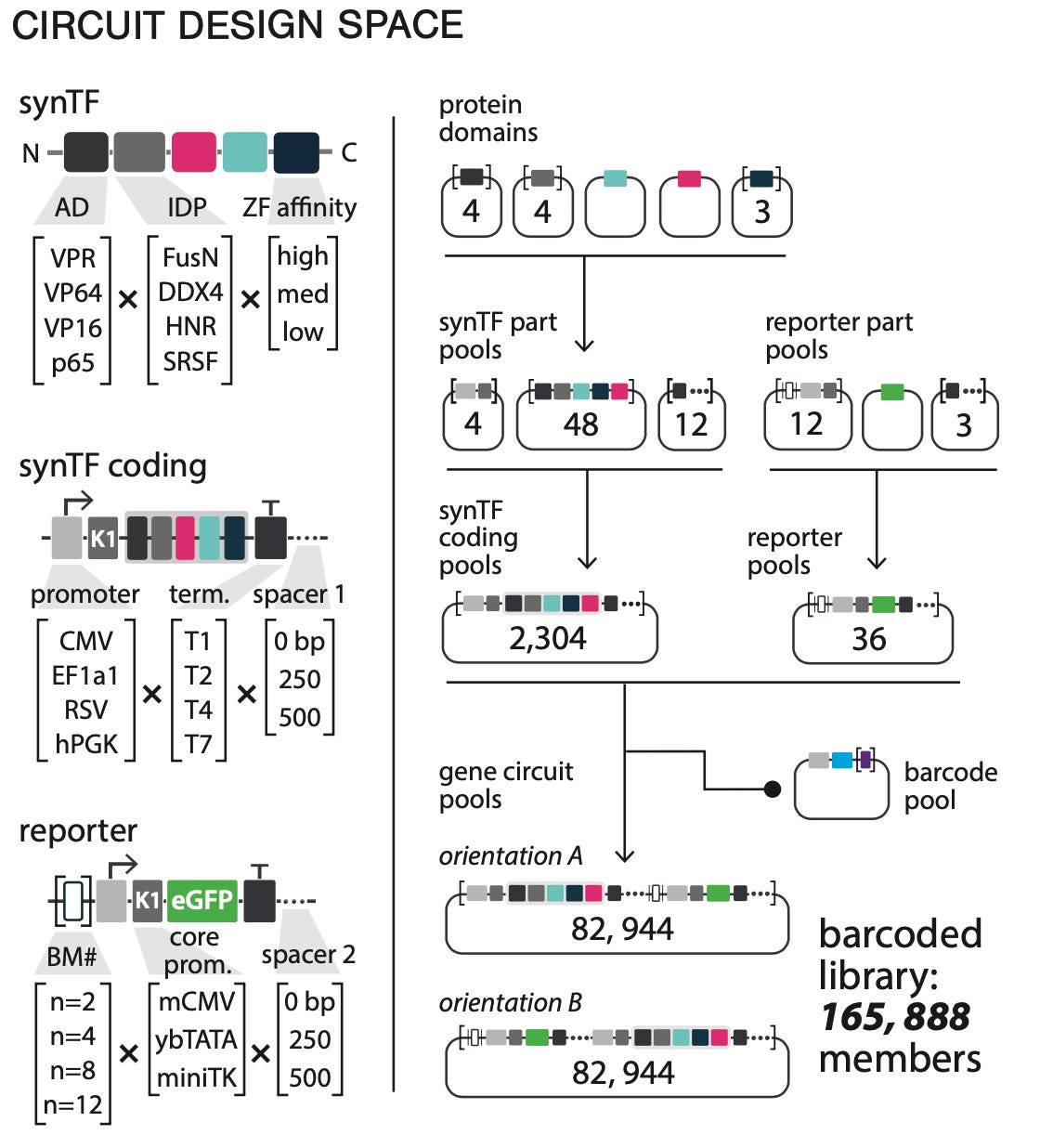
Their pooling screening approach was able to assign barcodes to 95.3% of that total design space. That’s a lot of circuits. It’s worth pausing here to reflect on the value of pooling experimentation using barcodes. In biology, there are broadly two ways to scale experimentation. The brute force approach is to simply scale the number of experiments you run using process automation like robots and computer vision. Alternatively, you can increase the number of distinct things you can measure in a single experiment. This second approach is where genomics shines, and this is a great example of it.
Learning models of genetic composition
As the scale of single-cell genomics data exploded, scientists like Aviv Regev have been able to greatly expand their modeling efforts. This is the foundational vision behind the Human Cell Atlas, which is an attempt to create the Google Maps for human cell types—a rich and interactive resource linking gene expression programs to cellular phenotypes.
Similarly, the scale of the circuit data that can be collected with a system like CLASSIC opens the door to a new set of modeling approaches. This study used the large-scale composition-to-function data to fit a random forest model of circuit behavior:

As our models of individual molecules become more robust, we’re now starting to learn the rules of how they work together. Let’s go galaxy-brain for a second. Here’s one of the coolest ideas from a closing paragraph of a paper I’ve read in a while:
While extensive recent work has used ML approaches to develop sequence-to-function models for various classes of genetic parts our work serves as a starting point for developing AI-based models of gene circuit function that use part compositions as learned features. While our current work has focused on mapping a design space of 10^5 compositions, it may be possible to create predictive models for more complex circuits with far more expansive design spaces by using data acquired with CLASSIC to train high capacity deep-learning algorithms (e.g., transformers) which require much larger datasets than currently exist.
Currently, OpenAI is demonstrating to the world in real-time how the Scaling Laws for AI work. With more data, more compute, and more parameters, Large Language Models like GPT-4 start to demonstrate incredible emergent capabilities, like scoring in the 90th percentile on the Uniform Bar Exam.
I’m not sure why this won’t also be the case for synthetic biology. We already have powerful models of specific parts. Now we’re learning models of parts composed together into circuits. What about models of combinations of circuits—getting us closer to predictive models of whole cells? Or combinations of cells—getting us closer to models of tissues?
Can we compose the outputs of our BioML models together like a set of Matryoshka dolls? If we can, it will probably let us design new biological systems more quickly—both for new medicines, and for industrial applications.
All en route to a flourishing digital and biotic future.
Thanks for reading this essay about exciting new work from the frontier of genetic circuit design. If you don’t want to miss upcoming essays, you should consider subscribing for free to have them delivered to your inbox:
Consider joining me at SynBioBeta this year to learn more about what else is happening in the world of synthetic biology. You can use the code H7TGP_JC10 for a 10% discount.
As always, thanks to my wonderful editor Kelda.
Until next time! 🧬
Clearly, the writing habit has been hard to kick. Life works in funny ways.
Note, the GPT-4 scores are suspicious, it's possible that the scores reflect it regurgitating/rephrasing answers that were in its training data, not actually generating correct answers it has never seen before.
(It can definitely sometimes give correct answers to questions it has never seen, but not reliably. The paper claims to check for data contamination in their testing, but the procedure they describe seems weak to me. They look for substring matches of the questions, and I expect the internet will have a lot of "the test asks something like X and here's the answer" to evade test-makers searching for test cheatsheets. Also, someone tried to reproduce the Leetcode scores reported by OpenAI. GPT-4 is good at Leetcode problems posted before the training cutoff date, and bad at problems posted after.)
Yee Haw to all this!!