
Scaling CRISPR Cures
Realizing the potential of genetic medicine

Welcome to The Century of Biology! This newsletter explores data, companies, and ideas from the frontier of biology. You can subscribe for free to have the next post delivered to your inbox:
Enjoy! 🧬

With modern tools, a small molecule drug discovery campaign often starts by screening millions—or even billions—of distinct chemicals. The same is true for antibodies. Blockbuster drugs like Humira that are sifted out of these enormous “libraries” of candidates are very lucrative needles in a molecular haystack.
For CRISPR medicines, things are a bit different.
Nearly identical CRISPR medicines can be reprogrammed to treat entirely new diseases. Let’s consider a pair of examples.
In June of 2021, Intellia Therapeutics, in partnership with Regeneron Pharmaceuticals, dosed six patients suffering from a rare genetic disease called transthyretin amyloidosis (ATTR) with an experimental medicine called NTLA-2001. This medicine consists of three distinct parts: an mRNA molecule encoding the Cas9 enzyme, a guide RNA molecule that targets Cas9 to the transthyretin (TTR) gene, and the sphere of lipids that encapsulates them called a lipid nanoparticle.

When delivered through a vein, the nanoparticles and their cargo circulate through the blood to the liver—where they are swallowed up by liver cells. Once inside of cells, the Cas9 mRNA is translated into the Cas9 enzyme and forms a sequence-specific editing complex with the guide RNA.

Transthyretin amyloidosis is caused by the accumulation of mis-folded TTR proteins which can lead to nerve and heart failure. In the case of NTLA-2001, the guide RNA programs Cas9 to insert mutations into the TTR gene—causing it to be effectively silenced. When patients were dosed, they experienced up to a 96% reduction in the levels of TTR proteins in their serum when compared to their diseased baseline levels.
Three years later, Intellia dosed ten patients suffering from an entirely different genetic disease called hereditary angioedema with another experimental medicine called NTLA-2002. Patients with hereditary angioedema experience painful swelling attacks that can be fatal if they occur in the airway and block breathing. NTLA-2002 was designed to target the kallikrein B1 (KLKB1) gene, which encodes a critical step in the molecular cascade that leads to the swelling attacks. The results were remarkable: “Among all the patients, the mean percentage change in the number of angioedema attacks per month from baseline through the latest assessment was −95%.”
NTLA-2001 and NTLA-2002 target different genes in different pathways that cause different diseases. But they are nearly identical. The lipid nanoparticles are the same. The Cas9 mRNA molecule is the same.
The only difference between the medicines is 20 nucleotides in the guide RNA.
Unlike small molecule drug discovery or antibody development, designing these critical 20 nucleotides is not a massive and intractable search problem. It can be done on a laptop computer, and the designs can be verified with DNA sequencing.1
For blood, eye, and liver diseases, there are already established delivery technologies and entire clinical protocols in place for testing new medicines. As we’ve seen, new gene-editing technologies are being invented—and even AI-generated—on a continual basis. Labs and companies around the world are hard at work expanding the set of tissues that these systems can be safely delivered to.
This is why Fyodor Urnov, the widely cited Berkeley scientist responsible for coining the term “genome editing” back in 2005, calls CRISPR “a platform for cures.” In theory, we could scalably test and produce these cures—even for patients with mutations found nowhere else in the human population—for every blood, eye, and liver disease with a genetic basis. Over time, with second-generation tools and delivery technologies, CRISPR could be used to treat the 400 million people worldwide suffering from genetic diseases—and could even extend into the realm of complex diseases.
So why aren’t we doing that?
This is the question that Urnov has been asking for many years, and the answer is complicated. Currently, there is a multifaceted thicket of regulatory, economic, and technological challenges preventing this vision from becoming a reality.
Instead of focusing on scaling the infrastructure necessary for personalized medicines, CRISPR’s translation into the clinic is being pursued in a business-as-usual approach by a finite set of for-profit companies. Each of these companies is focusing on diseases with economically justifiable markets, which has caused substantial crowding around a small set of blood disorders such as sickle-cell disease and beta thalassemia.
And it hasn’t been smooth sailing. As the biotech sector has struggled to reset after the “sugar rush” of funding that flowed into the market during the COVID pandemic, nearly every public CRISPR company has undergone rounds of layoffs.2
When medicines do make it into the clinic, they are expensive. Casgevy, the first FDA-approved CRISPR-based therapy, costs 2.2 million dollars. While there is a real pharmacoeconomic argument to be made that these medicines will save the medical system money in the long run by replacing a lifetime of complex medical care for very sick patients, it makes access difficult in the near term and causes healthcare spending to become much spikier.
Every time one of these million-dollar gene therapies gets approved, Peter Marks, the director of the Center for Biologics Evaluation and Research (CBER) within the FDA, gets an email from the agency’s commissioner, Robert Califf. As Marks puts it, they usually have “a lot of exclamation points.”
Urnov—now the Director of Technology and Translation at the Innovative Genomics Institute (IGI) founded by CRISPR Nobel laureate Jennifer Doudna—isn’t content with the status quo either. Recently, the IGI launched a new collaboration with Danaher aiming to “scale up the development of CRISPR cures.”
The ambitious vision is to lay the foundation to go from “one to a thousand” new therapies. Doing this will be incredibly challenging. We’re going to explore why the system currently works the way it does—and the problems they will need to solve in order to change it.
But first, let’s imagine how it could work if they succeed. As Urnov’s Lennon-inspired article asks of us, we’re going to imagine CRISPR cures.
CRISPR On-Demand
In a 2021 opinion article in Molecular Therapy and a subsequent New York Times article in 2022, Urnov laid out a vision for what a future with ubiquitous CRISPR medicine could look like if we work for it.
For longtime readers of this newsletter, it’s a clear 4-S story. The advances made in DNA Sequencing and Synthesis, in combination with new Software tools, could be used to deliver personalized medicines on an entirely new Scale.
To start, we could sequence the genome of every patient. Even this first statement would have been science fiction at the turn of the century. We had only just completed the Human Genome Project, which took over a decade and cost billions of dollars.
Consider how quickly technology has improved even since Urnov wrote this article three years ago. In 2022, researchers at Stanford broke the world record for the fastest genetic diagnosis, sequencing a patient’s complete genome in five hours and two minutes. With renewed competition in the sequencing market, the race is on to deliver complete genome sequences for $100 or less.3
For patients suffering from Mendelian disorders where a single gene is mutated, this can produce an immediate diagnosis. Next, the gene would be visualized in a genome browser where its full sequence could be collected.
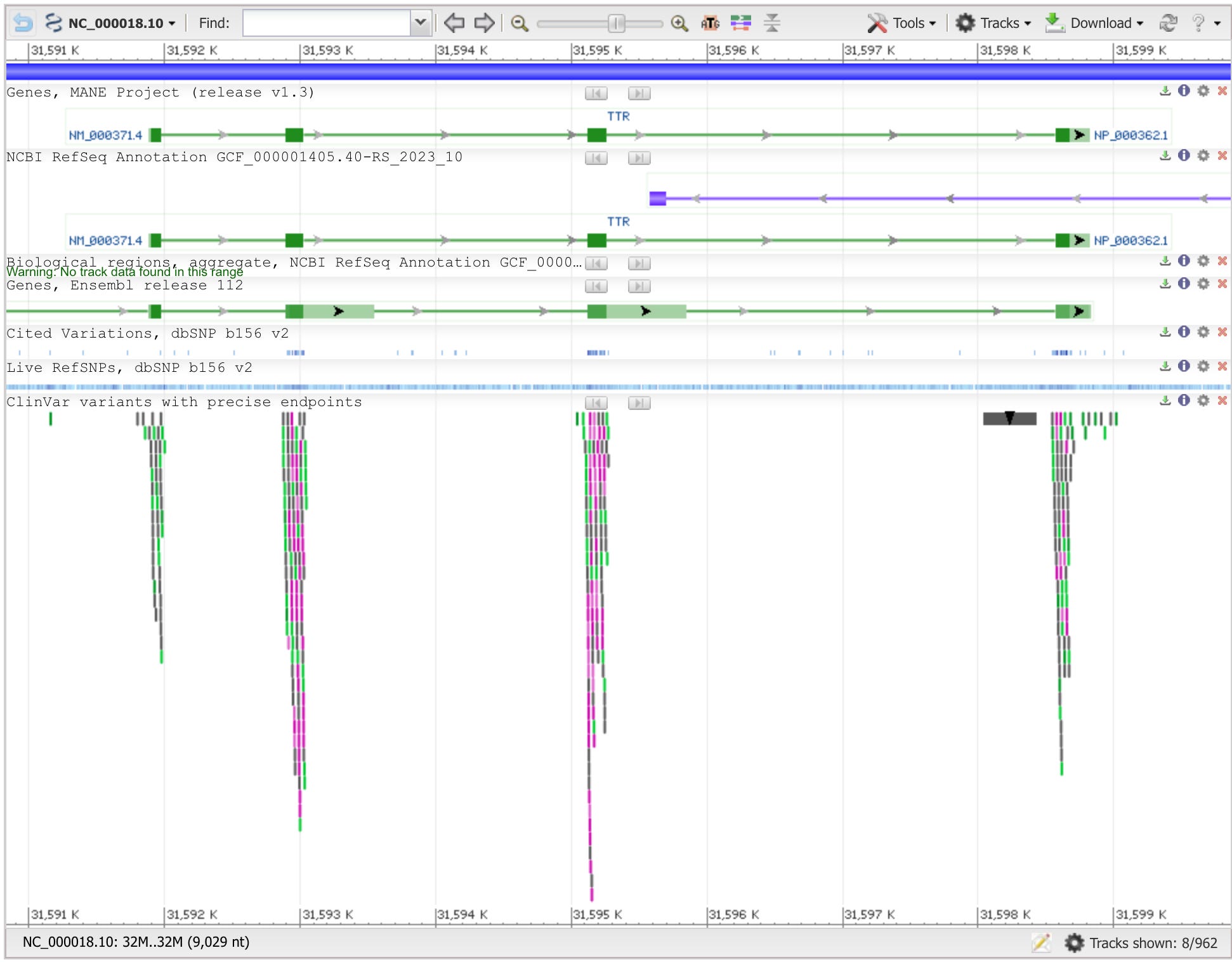
With the gene sequence in hand, a scientist or physician could run the same standard software tools that Intellia used to design their guide RNAs and screen them for potential off-target edits. The guide RNAs could then be ordered online from a commercial provider. Turnaround times are typically days, not weeks. In the not-too-distant future, machines for synthesizing the guides could be distributed in hospitals around the world if DNA printers continue to improve and are adapted to produce RNA.
For patients with severe need and no other option, this could be all that is required to produce a cure.
The guide RNA would be packaged into a delivery vehicle, and the patient would be dosed with their personalized therapy. Sequence. Design. Print. Dose.
As the software tools for CRISPR design continue to improve and we accumulate more data, this could prove to guarantee sufficient safety. For additional certainty, we could develop protocols for using simple blood samples and DNA sequencing to verify on- and off-target editing with a patient’s own genome.
What if CRISPR becomes so safe and standardized that we use it to treat additional diseases?
Consider statins. Hundreds of millions of people take these small molecule pills to manage their cholesterol levels and risk of heart disease. They are one of the most commonly prescribed drugs in the world. But if Verve Therapeutics succeeds in achieving their goals, that may no longer be the case.
Verve is currently developing a CRISPR therapy for a rare form of heart disease, but the company’s long-term vision is to expand their efforts to also treat the more common type of heart disease. As Urnov argues, in the future, “one gene edit could replace daily medications such as statins.”4

Clearly, this is a very different future from the present. It’s a world where every patient’s genome is routinely sequenced, and personalized genetic medicines are as commonplace as small molecule drugs.
So, what’s preventing this from happening? Let’s examine the first major roadblock in the current system: the time and cost associated with clinical development.
New Drug, New Trial
Modern clinical trials are a fairly recent invention. Up until the turn of the 20th century, there was effectively no regulation for the development and sale of new drug products. The FDA only came into existence in 1906 and gradually evolved into its current form after two major belt tightenings in 1938 and 1962.
Now, new drugs are brought to market through an intensely standardized and regulated gauntlet of preclinical and clinical phases.
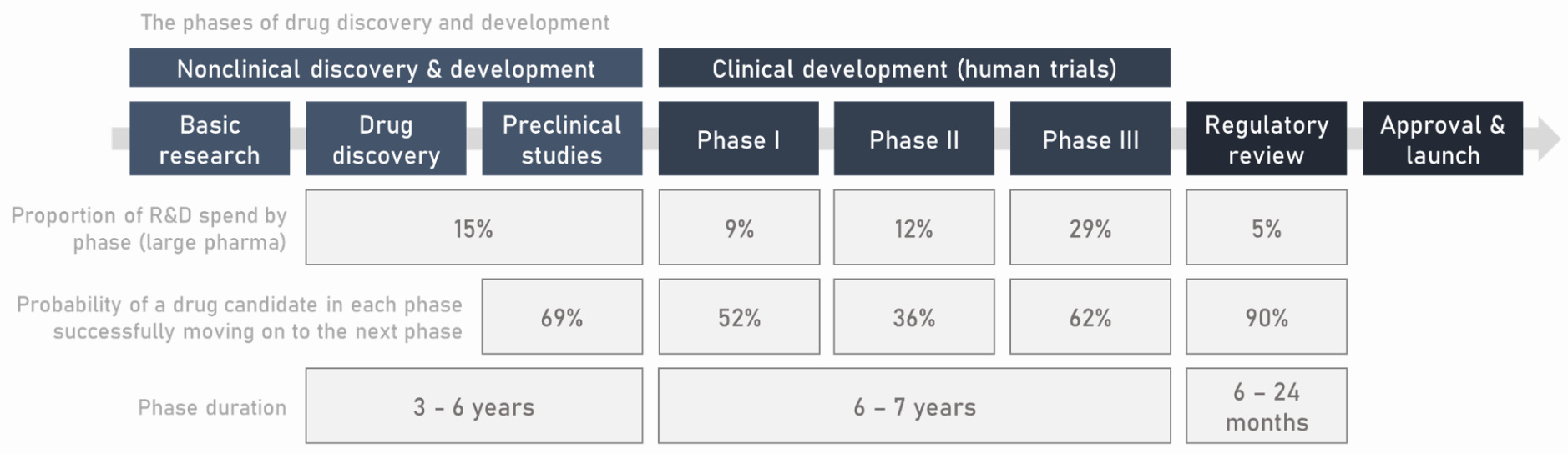
These regulations were largely successful in their goal of increasing the burden of evidence required to market new drugs and make medical claims about their benefits. Understanding the full consequences of this system—both good and bad—and why modern trials have gotten so expensive is a massive topic.
But when it comes to scaling CRISPR cures, there’s one particularly important detail to know:
No matter how similar two drugs are, the entire set of preclinical and clinical phases of development need to be repeated. New drug, new trial.
By Urnov’s estimates, this entire process would be “a four-year journey likely to cost at least $8 million to $10 million.”5 This makes the development of personalized CRISPR treatments effectively impossible. No company can justify these types of costs for rare—potentially even n-of-1—mutations, and patients often don’t have four years to wait to be cured.
The current regulatory framework was established in an era when nearly all new drugs were small molecules—before even antibodies were a class of medicine. From this perspective, it made a lot of sense to treat every new “test article” as its own unique entity. Even minor changes to the structures of small molecules can dramatically alter their impact on human physiology. In medicinal chemistry, there is a saying, “Change a methyl, change the drug.”
But is this true for CRISPR?
Are NTLA-2001 and NTLA-2002 really completely different medicines because of the twenty differing nucleotides in their guide RNAs? The answer is likely “no,” and Peter Marks and the FDA appear to agree.
On May 29th, the FDA issued a new draft guidance for a Platform Technology Designation Program to ease the regulatory burden for new drugs that use the same underlying technology. As Peter Marks put it, “We have to be thinking now because if every base pair changes we're going to have people come back to the agency, there are a lot of nucleotides out there that can go wrong in the genome. And I don't think we can have 3 billion visits to the FDA a year, so we'll have to figure this out.”
This is a really big deal. In this new potential framework, a company could recycle data between drug programs. We already know how the lipid nanoparticles and mRNA molecules behave in humans. Instead of reproving this, the focus can shift to proving the safety of the new guide RNA. As Jorge Conde of a16z put it, “We may be entering the 'reusable rocket' era for biotech.”
To fully understand the ramifications of this proposed regulatory update, let’s consider the current economic incentives of drug developers without any ability to rapidly reprogram existing medicines.
Producing Blockbuster Hits
In the world of tech investment, returns follow a steep Power Law distribution. Nearly every new startup fails, but a small handful of massive outliers manage to make up for this fact. Strangely, venture capital is one of the best-performing asset classes despite having the highest failure rate for any given investment.

When it comes to drug development, the Power Law may be even steeper. Not only do most drugs fail to gain approval, but most approved drugs still fail to recoup their development costs. Alex Telford describes this phenomenon nicely, saying, “This extreme unevenness of potential returns means that pharmaceuticals is fundamentally a hits-driven business. Because drug development is so costly and most launches are duds, the industry as a whole relies on blockbusters—drugs with annual global revenues of $1 billion or more—to cover the flops and keep the entire enterprise afloat.”

Historically, producing these types of blockbuster hits required developing treatments for common diseases with large patient populations. Lipitor—one of the most widely sold types of statins that we looked at earlier—broke records in the early 2000s for its multi-billion dollar sales.
Over time, the pharmaceutical industry’s focus has shifted towards rare diseases for producing new blockbusters. This shift can largely be attributed to three primary drivers:
Regulation. The U.S. passed a new law called the Orphan Drug Act of 1983, aiming to incentivize drug developers to make drugs for underserved patient populations. By and large, the incentives worked.
Increasing R&D costs. Clinical trial costs have steadily swelled over the years, disincentivizing development for drugs that would require larger cohort sizes to gain approval. Costs per patient have climbed from $10,000 up to as high as $500,000.
Pricing evolution. Biopharma companies tested the waters with new strategies, charging massive prices for small patient populations. The most extreme example so far is Hemgenix, which is a $3.5M gene therapy for Hemophilia B—which has less than 300,000 occurrences worldwide.6
As Matthew Herper incisively described it,
Instead of dealing with the difficulty of collecting data on new medicines, both society and government sidestepped it by focusing on treatments for much rarer illnesses: relatively rare cancers, rheumatoid arthritis, and multiple sclerosis to name a few, not to mention very rare illnesses such as cystic fibrosis or paroxysmal nocturnal hemoglobinuria.
The current “goldilocks zone” for new blockbusters is the set of rare diseases that are not too rare—the equation is still X patients times Y price >= $1B; even with a large Y, X still needs to be big enough—have sufficient unmet need, and have well-described underlying mechanisms that can be addressed by existing technologies.
These are the constraints that the current CRISPR companies are working with, and their clinical pipelines reflect this.
In the future that Urnov dreams of—and that the FDA is increasingly sympathetic towards—this all could change. Imagine if the draft guidance for platform approvals is passed into law. A company like Intellia could gain approval for platforms rather than products. The fundamental technology underlying NTLA-2001 and NTLA-2002 could be approved as a platform for curing genetic diseases of the liver, dramatically reducing the time and development costs associated with curing any one disease.
What if organ-level platform approvals for programmable medicines became the new hot blockbuster category?
But for any of this to come to fruition, there’s a looming technical bottleneck: we’ll need to solve the manufacturing challenges associated with producing personalized genetic medicines on demand.
The Last Mile of Manufacturing
Not all drug modalities are equal. Different types of medicine—such as small molecules, biologics, and cell and gene therapies—have different tradeoffs in terms of clinical benefit, trial risk, and manufacturability. Small molecules like Lipitor aren’t curative but can be cheaply and abundantly produced. Cell-based therapies like CAR-T can cure some cancer patients but are a nightmare to formulate. Biologics like Novo and Lilly’s GLP-1 agonists have caused enormous headaches when attempting to scale to large patient populations.
If personalized gene-editing medicines like CRISPR are ever to become as commonplace as small molecules, they’ll need to become equivalently straightforward to make.
There are good reasons to think that this is possible. If these medicines—like the ones produced by Intellia—are simply formulations of lipids and RNA, the COVID mRNA vaccines provide an existence proof that drugs with identical nucleic acids can be scaled to dose billions of people. Seemingly, the major manufacturing hurdle will be producing drugs with different nucleic acids for different patients. This is why it’s so interesting that Danaher is the company collaborating with the IGI.
Danaher is the 800-pound gorilla of biomedical tools and manufacturing. As a ~$200B conglomerate company, Danaher has steadily acquired many of the leading infrastructure providers for the life sciences ecosystem. In 2018, they acquired Integrated DNA Technologies, Inc. (IDT), which is one of the leading providers of synthetic DNA and RNA—including CRISPR guide RNAs. In 2021, they acquired Aldevron, a major manufacturing provider for nucleic acids, proteins, and biologics—including “High-quality, scalable mRNA manufacturing meeting a wide range of requirements from linear template through LNP formulation & encapsulation.”

With all of these different pieces, it’s hard to think of a company better equipped to tackle this challenge. I’ll be curious to see what this Danaher and IGI initiative is able to achieve in potentially vertically integrating these different technologies into cost efficient systems that can be more widely adopted by hospital systems.
Thinking back to our future scenario with ubiquitous CRISPR cures, there will also need to be continued adoption of genome sequencing and rapid genetic diagnoses. Again, with turnaround times already being measured in hours rather than days, this seems like more of a question of achieving widespread deployment rather than needing further technical leaps.
We’ve now looked at the regulatory, economic, and manufacturing challenges associated with scaling CRISPR cures. In each case, there is a reason to be optimistic that the future will be better than the present. Let’s zoom back out to see what all of this could lead to in the future.
Putting the Pieces Together
CRISPR therapies—along with other types of genetic medicines—are a step towards making programmable cures for disease. By changing twenty nucleotides in a drug for transthyretin amyloidosis, Intellia designed a potentially curative treatment for hereditary angioedema. This is not how drug development typically works.
But despite the promise, bottlenecks in the existing drug discovery ecosystem have rate-limited the scalability of these approaches. The same companies that developed these potential “platforms for cures” are laying off employees from their research teams. Regulatory hurdles established for a prior generation of technologies have come to dictate the business strategies of drug developers. We still can’t produce genetic medicines with the same speed and cost as small molecules.
Even in the face of these setbacks, there are many reasons to be optimistic that the future of these technologies is bright. Danaher and the IGI—world leaders in the manufacturing and development of CRISPR medicines, respectively—have joined forces in a new initiative to go from “one to a thousand” cures for genetic disease. Just this week, the FDA released a draft guidance indicating that we could soon operate in a world where platform technologies have accelerated pathways to develop medicines for drugs outside of the current “goldilocks zone” where drug developers are hunting for the next blockbuster drugs.
Where could things go in the future?
In the near term, these technologies and new regulations are likely to have the biggest impact on ultra-rare diseases. But as if this essay wasn’t already speculative enough, let’s dream a little bit bigger. A lot of new biomedical infrastructure will need to be built to scale the development of personalized medicines for genetic diseases. What if these new ultra-rare cures are like the pricey Tesla roadster that paved the way for the more broadly affordable Model 3?
Once the infrastructure is in place for rapid and inexpensive testing and dosing of n-of-1 medicines around the world, shouldn’t that drive down the costs of clinical development for all subsequent genetic medicines? Consider one of Alex Telford’s predictions for a new source of blockbusters in the future:
The biotech toolkit (incl. gene therapy) expands into more common acquired diseases as our understanding of complex biology improves and manufacturing costs decline. This leads to a revisiting of highly-genericized out of favour indications with remaining unmet needs (e.g. cardiovascular)
Already, we’re seeing companies like Verve pursue this strategy. In a world where companies like Danaher—or startups with new manufacturing technologies and solutions—offer a cheap and globally distributed infrastructure layer for any company with a compelling new product to build on top of, we could see a new golden era of investment in cures for more common diseases.
If even part of this vision becomes reality, the future of medicine could look very different than it does today.
Thanks for reading this essay about scaling the development of programmable genetic medicines.
If you don’t want to miss upcoming essays, you should consider subscribing for free to have them delivered to your inbox:
Until next time! 🧬
Specifically, the study describing NTLA-2001 cites the Cas-OFFinder algorithm, GUIDE-seq, and SITE-Seq as key tools in their design workflow.
Editas cut back on their headcount and clinical programs in January last year, Beam Therapeutics laid off employees in October last year, CRISPR therapeutics laid off employees in November last year, and Intellia laid off staff—including members of its early-stage research team—in January this year. This list is non-exhaustive.
One technical point: normally, when people talk about personal genome sequencing, they are primarily generating whole-genome sequencing reads, and aligning the reads to existing “reference genomes.” With new algorithms for automated genome assembly and improvements in sequencing technology, in the future everybody could have their own genome assembled completely from scratch.
Verve had a slight clinic setback in April, which they attribute to the LNP delivery vector they used. They are now enrolling patients for a new version of their therapy with a different vector. This highlights the challenge of CRISPR delivery which is still far from being a solved problem. The mechanism by which LNPs deliver their cargo is inherently inflammatory. It will be important to continue to improve LNP designs and to develop other alternatives.
This estimate is just accounting for the experiments required to start a clinical trial, and to establish a manufacturing protocol for making the treatment.
Hemophilia B has an incidence of 1 in 30,000 males, and largely doesn’t impact women.
Man, how amazing is Jennifer Doudna? First she invents the world-changing CRISPR technology, then when it's use to drive significant human results gets stymied by regulation and the positive non-academic impacts of CRISPR stall for a decade, she takes on the regulatory Beast and gets the Platform Technology Designation? A towering role model and hero for us all.
Amazingly cogent, comprehensive, and useful summary of the landscape too, sincere kudos for putting this all together and publishing it!
Another very well written article. I’d be interested in seeing the evolution of pricing for such curative medicines if the regulatory path follows the platform approach indicated by the FDA.