
Generating (Somewhat) New Biology with AI
Using protein language models for Digital Evolution

Welcome to The Century of Biology! This newsletter explores data, companies, and ideas from the frontier of biology. You can subscribe for free to have the next post delivered to your inbox:
Enjoy! 🧬
After roughly four billion years of stumbling search, Evolution has churned out an absurd diversity of organisms. Microorganisms that can convert light into energy, trees that grow to hundreds of feet in height, insects capable of farming other insects, and mammals capable of forming technological civilizations. All of this diversity is encoded in genetic information, which primarily produces a universe of molecular machines—proteins.
If the scale of organismal diversity is absurd, the universe of proteins is practically incomprehensible. The millions of unique species on Earth collectively produce somewhere around a trillion different distinct proteins. In this enormous set, there are some very pragmatic proteins responsible for building and maintaining cells and replicating DNA, but there are also more exotic machines, like the bi-directional nano-scale motors that bacteria use to navigate towards chemicals.
Still, evolved proteins represent an infinitesimal fraction of the total theoretical space of possible proteins. Consider the following thought experiment: how many possible proteins with 200 amino acid residues are there? Since there are 20 possible amino acids for each position, the answer is 20 raised to the power of 200. This number is so big that it makes the number of stars in the observable universe look small.
One of the most promising fields of biotech is de novo protein design, which aims to explore the full space of possible proteins for new types of molecular machines beyond what Evolution has already produced.
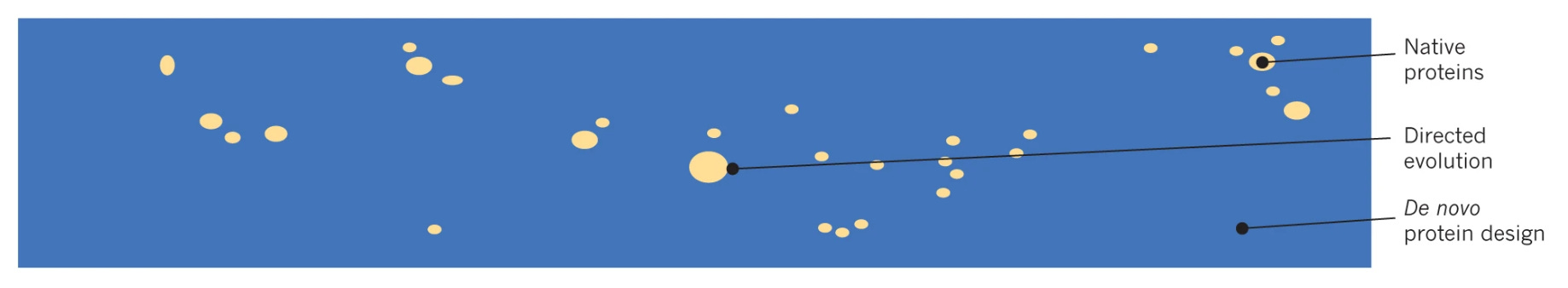
This is a really big idea. Directed evolution—the set of laboratory techniques for rapidly sampling new beneficial mutations to known proteins—was a Nobel-worthy idea that has already produced a lot of value. Sampling beyond known proteins opens up an entirely new world of possibilities.
Recently, de novo protein design has gotten a big boost from AI. Taking inspiration from the wave of exciting results in using AI to generate new text, images, audio, and video, scientists have retrofitted and repurposed the underlying algorithms to generate new proteins.
This isn’t just a big idea—it’s apparently a billion dollar idea. Last week, a new startup called Xaira Therapeutics announced its $1B Seed round of financing. David Baker, the senior author of the paper with the figure above, is a co-founder. While the vision for the company spans everything from basic biological research to clinical trial design, a major focus is to design new antibody therapies by improving recent generative AI algorithms from the Baker Lab.
So, when people say “generative AI for biology,” they are talking about the development of deep learning models capable of generating new types of biological outputs—like new DNA sequences or proteins. In the future there may be models trained on a variety of inputs capable of producing a variety of outputs, but right now, most efforts are focused on using specific algorithms for specific biological inputs/outputs.1

These models and their outputs have been getting a lot of attention. Last week, a bioRxiv preprint from Profluent Bio describing new AI-generated CRISPR proteins took the Internet by storm. And it wasn’t just my corner of the Internet with other biotech nerds—The New York Times covered the work the same day it was posted.
According to the NYT article, Profluent’s model “analyzes the behavior of CRISPR gene editors pulled from nature and learns how to generate entirely new gene editors.” I think this is close to true, but not quite right.
In a clever piece of detective work, Brian Naughton, a biological data science guru, asked a simple question: how distant are these new gene editors from multiple natural sequences? When lining up the new editors against naturally occurring proteins, he realized that he could “recreate 98% of the sequence (all but 24 amino acids) with 3 Streptococcus sequences.”

I don’t think Brian intended for this to be a dunk on Profluent’s work, and I don’t either. It would be effectively impossible for even the best CRISPR biologist to sit down at their keyboard and type out this new sequence, let alone confidently predict that it would actually work in cells. Clearly, in its exhaustive training on natural sequences, Profluent’s model is learning something really interesting about how CRISPR proteins work.
But the generated results aren’t entirely new. They are like one of Dali’s surrealist paintings, synthesizing disparate existing objects into a new unified whole. The novelty is in the composition, not the ingredients—which come from Nature.
One way to look at this is that it’s like Digital Evolution, where the AI algorithm is sampling from the space of all possible CRISPR permutations.

This makes a lot of sense when considering how these models work. To understand all of this, we’re going to look under the hood of this specific approach. This will help bootstrap intuition for what we should expect—and not expect—from this first wave of generative AI models for biology.
A Quick Primer on Attention
When analyzing the growth of scientific knowledge, there are often fundamental results that seed important branching points in our understanding or capabilities. For the current wave of AI progress, that critical node is a 2017 paper from Google entitled Attention is All You Need.
This paper described a new neural network architecture called a Transformer. At its core, a neural network is a big pile of linear algebra that learns the relationship (function) between inputs and outputs. These learned functions can be used to solve hard problems like language translation that are difficult—or impossible—to produce solutions for by explicitly writing code.

In theory, given enough time and training examples, even the simplest neural networks can learn to approximate nearly any function. But we don’t live in the world of theory, so AI researchers need to invent new architectures, which are specific combinations of the underlying linear algebra operations that converge on interesting solutions in a reasonable amount of time.
So in reality, one of the layers in a Transformer model look something like this:
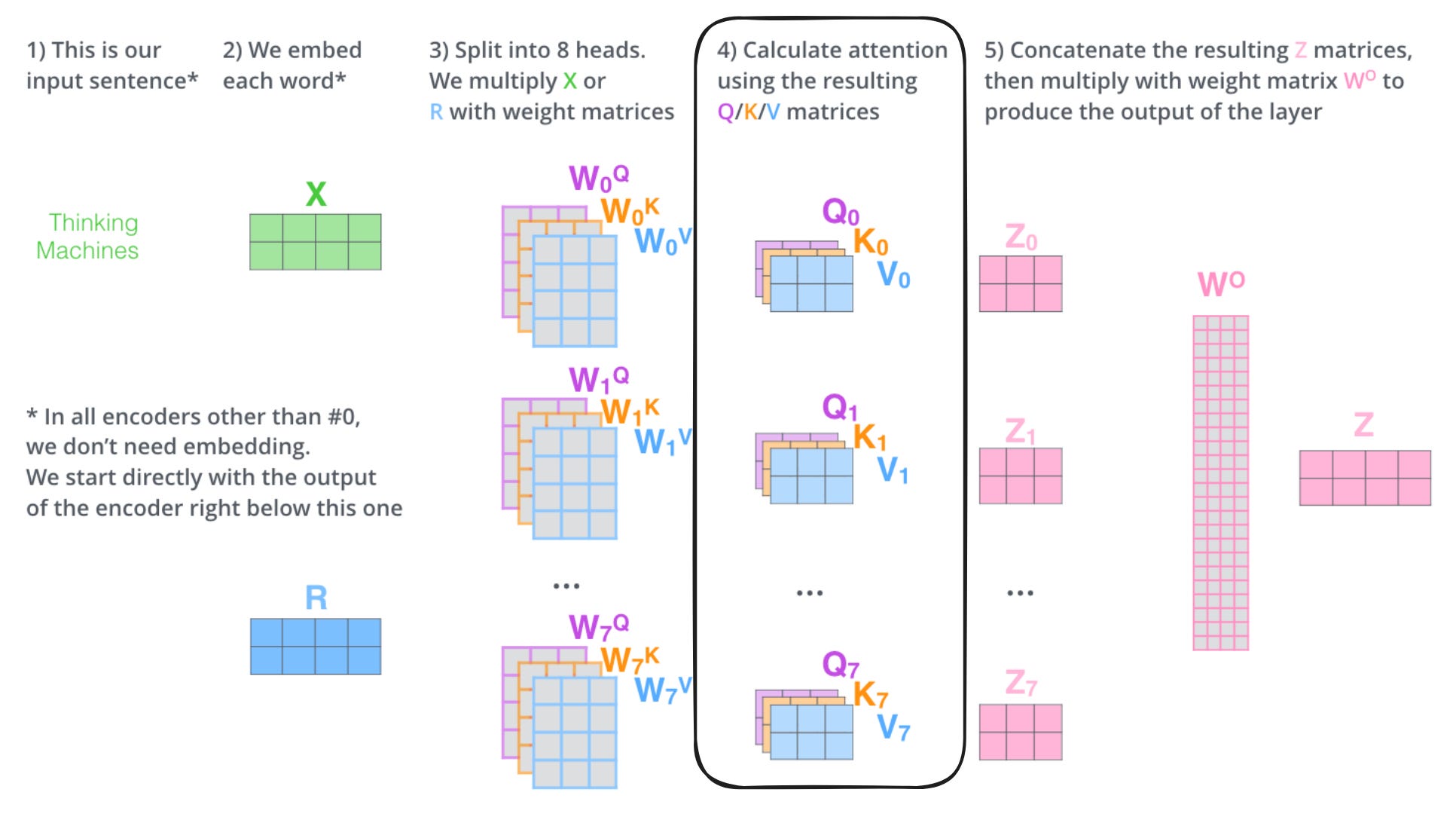
And much of the magic happens in the attention matrices (highlighted), hence the name of the paper. The query (Q), key (K), and value (V) matrices keep tabs on the relationships between each chunk of the input that is being processed. Visually, the attention patterns of a trained Transformer look like this:

Here, one of the attention heads—a specific copy of one of the matrices above—is detecting a relationship between “animal” and “it” in the sentence. Another is picking up on the relationship between “it” and “tired,” which is used to describe the animal later on in the sentence.
There’s an obvious question at this point: how do all of these matrices work together to actually learn that “it” is referring to “animal” in a sentence like this?
These matrices are parameterized by weights, which are just numbers that are multiplied with the inputs that can be tuned to improve the performance of the model. The training—or tuning of these weights—happens by computing the changes that would reduce the error rate based on known examples of inputs and outputs to the model.2
One of the ways to generate a lot of training examples for these networks is to mask parts of the input, and ask the model to guess the correct value that is hidden. This masking can be done a few ways. The mask can be applied in the middle of a sentence, like this:
 and Causal Language Models (CLM) in NLP | by Prakhar Mishra | Towards Data Science")
Or the last token can be masked, which can be used to train the model to learn the relationship between all of the preceding chunks of the sentence and what the next one should be. This is called causal masking.
 and Causal Language Models (CLM) in NLP | by Prakhar Mishra | Towards Data Science")
I realize that we’ve covered a lot of ground and that this explanation will be unsatisfying if you want to deeply understand Transformers. If you’d like to learn more, I’d recommend starting with the Illustrated Transformer article, or the 3Blue1Brown series on neural networks. Both are great starting points for building intuition about how these models work. They also both provide a nice rabbit hole of resources for further learning.
For now, here are the three takeaways we need to understand:
Neural networks learn to approximate complex functions, like language generation, by repeatedly tuning weights to perform better on training examples.
An important neural network architecture is the Transformer. These models have specific sets of matrices that track relationships between the elements of their input. This is called attention.
One of the best ways to train Transformers is to mask parts of the input and make the model correctly guess what is being hidden.
With these basics in mind, we’re in a better place to understand how these protein language models are generating new CRISPR sequences.
Generating (Somewhat) New Proteins
Biology and computer science have a unique bidirectional relationship. Studying biological computation gives us new ideas for how to program computers. The connections between biological neurons provided inspiration for artificial neural networks. Algorithms from computer science are also useful tools for learning new things about biology.

As the mountain of new machine learning techniques grows exponentially, cleverly repurposing these tools for biological purposes has become its own kind of art form. Around the time that language models started to really work, computational biologists started to have more success modeling proteins using these types of approaches.
Interestingly, many of the earliest results can be traced directly to new biotech startups. Surge Biswas, one of the lead authors of a 2019 protein modeling paper from the Church Lab, is now the co-founder and CEO of Nabla Bio. Alex Rives, one of the leader authors of a 2019 protein language model paper, is now the co-founder of EvolutionaryScale. And Ali Madani, lead author of a 2020 paper on protein language models, is the CEO of Profluent, the company that announced the AI-designed CRISPR proteins last week.3
In the 2020 paper and a pair of follow-up papers in Cell Systems and Nature Biotech in 2023, Madani and his colleagues developed and scaled causal language models for the task of generating new protein sequences.
Let’s think back to how this strategy works in the context of natural language. The causal training setup involves masking the last word in a string of words, and making the model guess what is based on the attention patterns in the rest of the string.4 Once the model is trained, this prediction step can be repeated to complete an entire sentence.
For example, the following prompt:
"Somatic hypermutation allows the immune system to"Could generate the sentence:
"Somatic hypermutation allows the immune system to be able to effectively reverse the damage caused by an infection."Instead of predicting the next word in a sentence, the model—called ProGen2—is predicting the next amino acid building block in a protein.

And just like a language model can be used to generate new sentences, ProGen2 can be used to generate new proteins.
This is how Profluent generated new CRISPR proteins last week. There’s only one more core detail we need to understand about their process: fine-tuning. The masking trick we’ve talked about is typically used as a pre-training step in developing Transformer-based Large Language Models (LLMs) for language applications. This step learns a general representation of language, before training a model on a more specific downstream task.
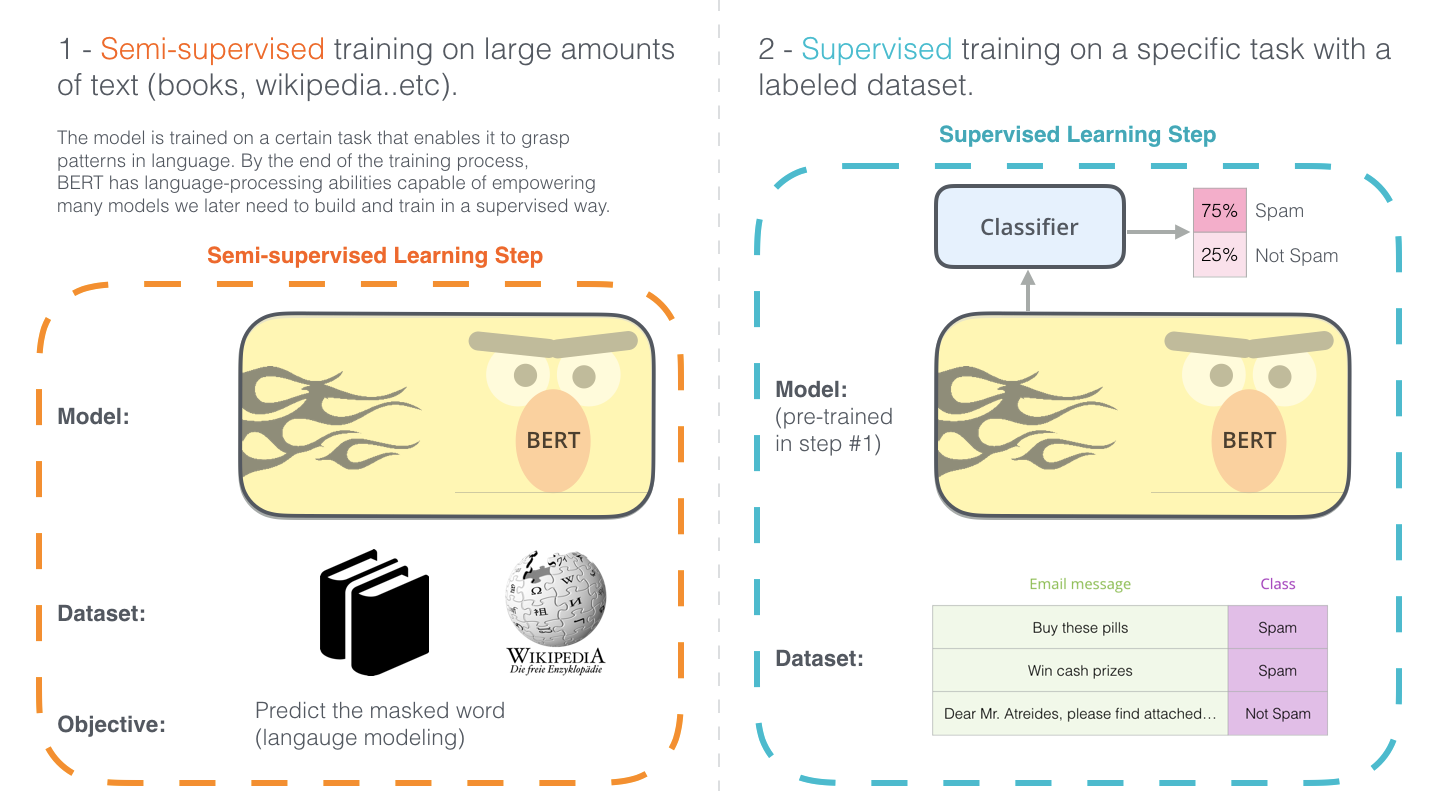
For proteins, ProGen2 was first pre-trained on a giant database of 500 million different proteins. To specifically design new CRISPR proteins, Profluent created two additional databases of examples of CRISPR proteins found in Nature for fine-tuning.5

The intuition here is that the pre-training process is learning deep principles of protein evolution, while the fine-tuning step is teaching the model to become the world’s expert in CRISPR biochemistry. It is tabulating the relationship between every pair of amino acids in every single CRISPR sequence that we know of.
With the final model, Profluent generated 4 million different versions of the CRISPR-Cas9 protein. Half of these were directly generated with no prompt. The other half were given prompts of 50 amino acids from different members of the Cas9 protein family.
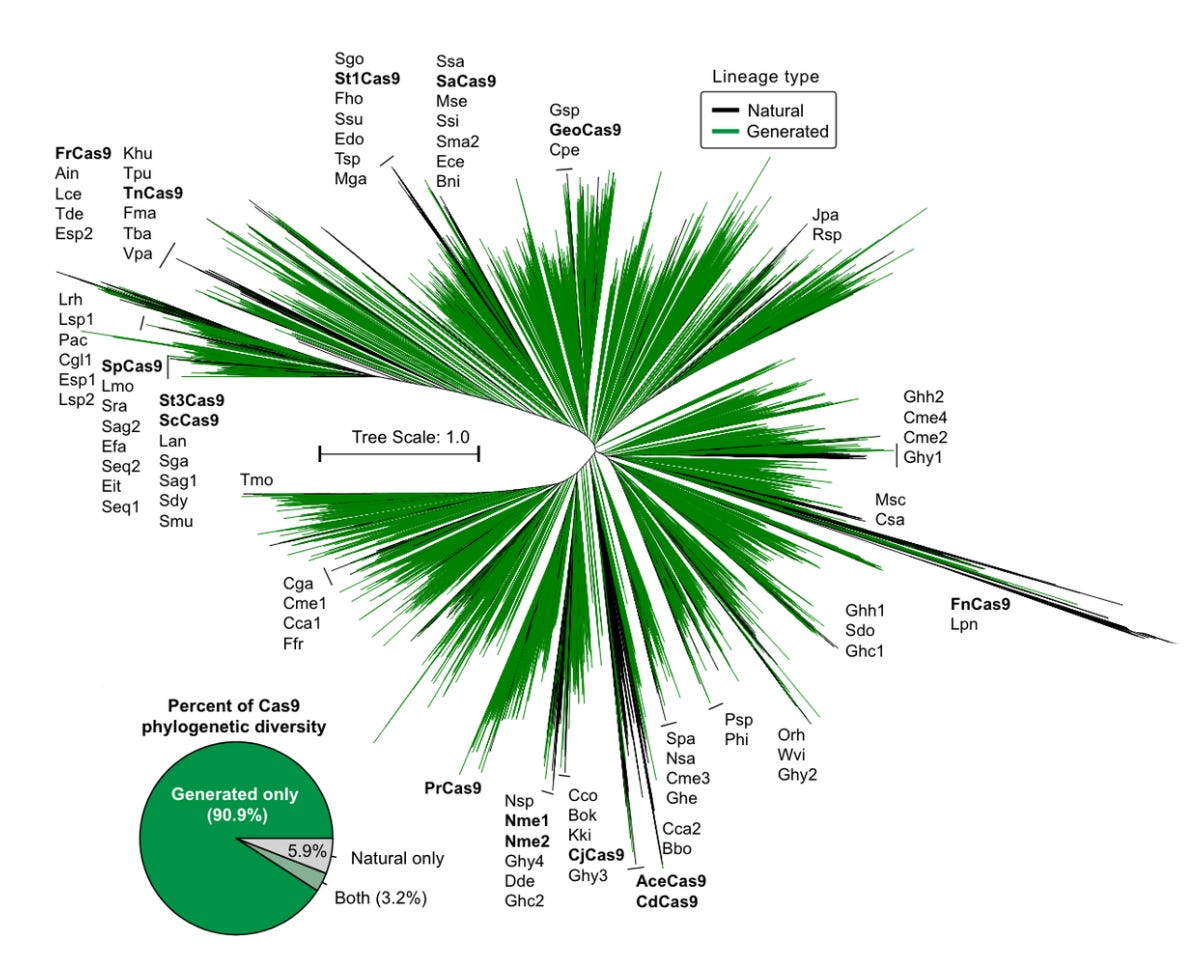
When put to the test in cells in the lab, some of the generated editors actually worked. Not only did they work, but they may be safer and more efficient gene-editing systems than spCas9.
But let’s revisit our earlier question: are these actually entirely new CRISPR proteins?
This is where things get fuzzy. One of the final generated designs, called OpenCRISPR-1 by Profluent, differs by 403 of its 1,380 amino acid residues compared to spCas9. That’s substantial. But when searching for related sequences, the closest result was 86.3% similar. And when comparing three or more sequences like Brian did, the number quickly approaches 100%.
When considering how the model is designed and trained, isn’t that exactly what we would expect?
Each time it adds a new amino acid to a sequence, the model is predicting the highest probability choice based on its knowledge of every known CRISPR. Again, this is more akin to Digital Evolution than it is to de novo design.
And that’s a good thing, because we currently don’t know how to actually design something as complex as CRISPR from scratch. In their introduction, the authors said that “for the design of complex functions as embodied by the gene editors in this work, structure-based approaches do not offer a straightforward solution. By contrast, language models provide an implicit means of modeling protein function (and thus structure) through sequence alone.”
So while these aren’t entirely new CRISPR systems—which is a tricky thing to really measure—this is an impressive demonstration of the generative capacity of protein language models.
They are powerful new tools in the growing computational protein design toolkit.
Final Thoughts
You may be wondering why Open-CRISPR-1 is named the way it is. It’s because you can download the sequence from GitHub and do whatever you want with it. According to Profluent, “OpenCRISPR is free and public for your research and commercial usage.” After filling out a simple Google form, the only requirement for use is to sign a simple non-exclusive license agreement.
I’m not a CRISPR patent lawyer—which is something I’m glad about—so I have no idea how this will hold up over time. If the CRISPR patent battles weren’t already messy enough, this adds an entirely new dimension: how do you prove that you computationally generated the new sequence and defend its novelty? This was the basis of an academic feud at MIT that dragged on for nearly four years.
Ultimately, I hope this CRISPR protein and it’s successors do open up access to gene-editing.
But as I mentioned when discussing other new tools for programming biology, there is still a suite of regulatory and financial challenges in the way of a future with ubiquitous programmable CRISPR medicines.
Even for companies that already have their own CRISPR intellectual property, it’s not all smooth sailing:
We could transition from modalities that require intensive development for every new application, to programmable modules where only the instructions need to be updated to make entirely new medicines.
This type of technology sounds like a cash printing machine. Surprisingly, nobody seems to have solved the business model problem for curing thousands of genetic diseases with CRISPR. Intellia, the company with some of the deepest expertise in this technology, had to lay off staff earlier this year.
Squaring these facts requires a consideration of the current economics of drug development. In an upcoming post, we’ll explore this in detail. Is there a platform strategy that could realize the full potential of CRISPR?
As the entomologist E.O Wilson once framed it, “The real problem of humanity is the following: we have Paleolithic emotions, medieval institutions, and god-like technology.” As we come to learn the language of Evolution and refine our capacity to steer it, we need to solve the complex human challenges that are rate-limiting broader access to better health.
Soon, we’ll dive into the details of how we might get to a “world where we have CRISPR on demand within weeks.”
Thanks for reading this overview of how protein language models were used to generate (somewhat) new CRISPR proteins.
If you don’t want to miss upcoming essays, you should consider subscribing for free to have them delivered to your inbox:
Until next time! 🧬
If there are already good examples of multi-modal models with better performance, please send these papers my way! One interesting recent example is the Evo model, which was trained on DNA, but was also used for RNA and protein prediction tasks.
This isn’t as complicated as it sounds. Consider linear regression. We have a simple linear predictor function of the form:
When fitting a linear model to best predict the output for a given input, we need to find the most optimal values for the coefficients (the Beta values). These coefficients are what ML researchers call weights, and just like for linear regression, there is an algorithm to do this estimation called back propagation, which is a bit more complicated because the number of weights is much larger.
One of the paper’s authors was Possu Huang—the first author of the de novo protein design manifesto mentioned at the beginning of this essay. It’s a small world.
In these models, the sentences are actually broken up into tokens, which might not exactly be words. For understanding, thinking about tokens just as words is a reasonable approximation.
One technical point: being able to train on DNA sequencing data instead of protein structure data is a big advantage for this class of models. Given the exponential cost decrease in DNA sequencing, the former is ubiquitous and easy to generate.
Firstly - your relatable prose + high intelligence is inspiring. I’ve been obsessed with these topics since turning down an opp to fund a series A in a de-novo startup. They went on to huge rounds and I’ve been catching up ever since. You deliver huge value and I can’t wait for the next one.
Even before the Evo model, genome scale language models first showcased the importance of the use of dna- based representations in capturing evolutionary trajectories of SARS-CoV-2. The important thing to note is that the model was first developed to be a foundation model trained on some of the largest and openly available prokaryotic gene sequences. GenSLMs are available at: https://github.com/ramanathanlab/genslm. The paper also provides ideas on the scaling of such models.