
Cellular shuttle systems
Engineering extracellular vesicles to deliver drugs

Welcome to The Century of Biology! This newsletter explores data, companies, and ideas from the frontier of biology. You can subscribe for free to have the next post delivered to your inbox:
Enjoy! 🧬

Our molecular armamentarium of nucleic acid therapeutics is constantly expanding in scope and complexity.
During the COVID pandemic, Moderna demonstrated the utility of working with a fundamentally programmable therapeutic medium by designing a novel vaccine within 48 hours of receiving the coronavirus genome. This type of nucleic acid programming is just getting started.
It’s easy to get overwhelmed by the growing set of acronyms—mRNAs, siRNAs, ASOs, ADARs, programmable RADARs, and CRISPR-based gene-editing systems are all valuable additions to our growing nucleic acid toolkit.
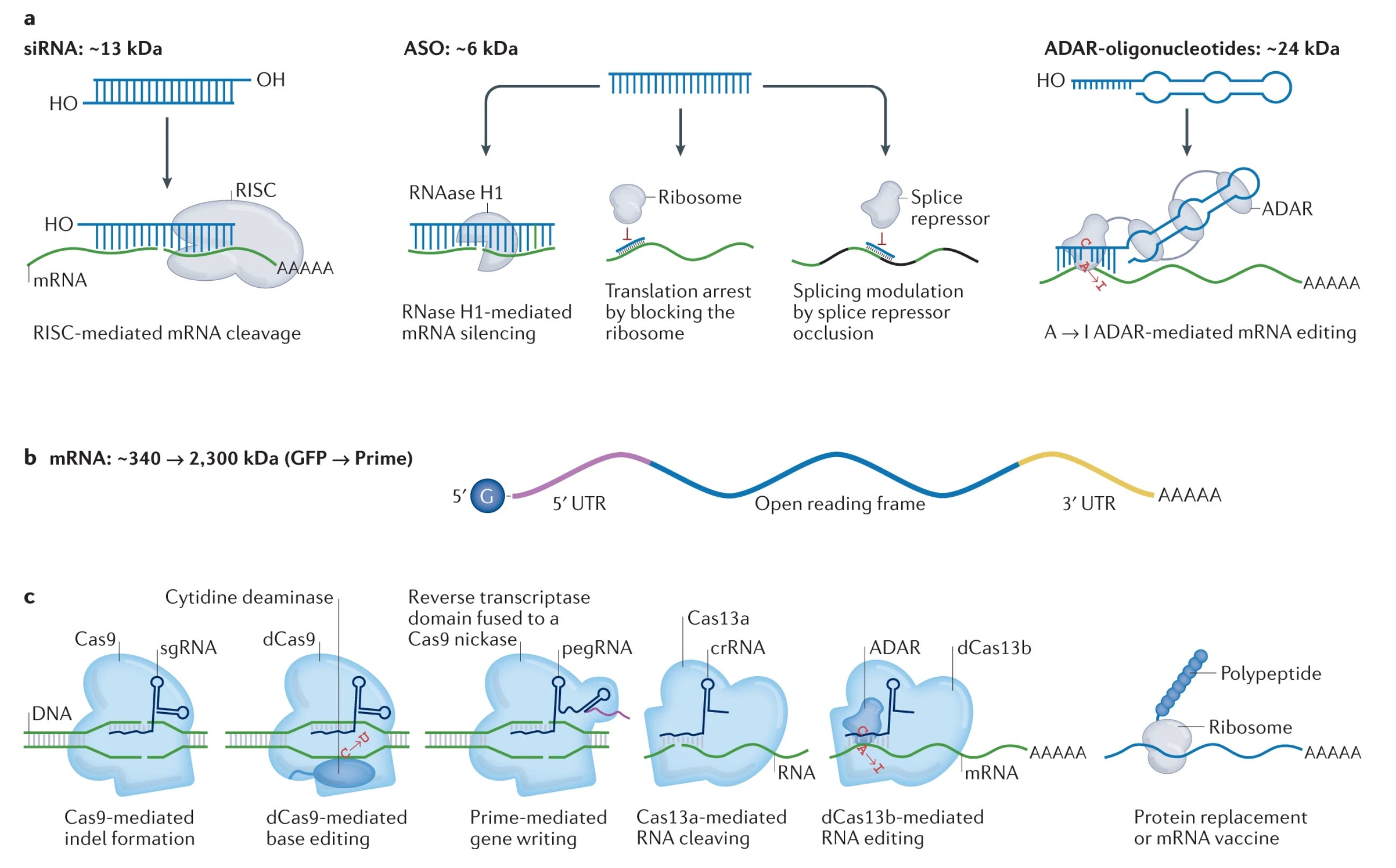
If you zoom out from the details of any specific mechanism, the core insight is beautiful: we are building tools to program cells. mRNA payloads program the production of proteins. Guide RNAs program the editing and writing of DNA. If DNA sequencing gave us the ability to read the language of cells, these tools give us the ability to write in it.
A major bottleneck for this paradigm is that we still struggle to deliver the messages we write.
Even predating RNA therapeutics, the delivery problem has historically plagued the field of gene therapy. In genetic diseases where we have complete knowledge of the disease mechanism, we can’t treat patients because of our inability to deliver a functional copy of the defective gene safely. To date, the biggest clinical wins for gene therapy have been in inherited retinal diseases where delivery is greatly simplified.

If we want a future where we can reliably program any cell in the body and we make genetic disease a thing of the past, we need to solve the delivery problem.
So, what are some of the candidate solutions?
At the highest level, delivery approaches are classified as viral or non-viral. It makes intuitive sense to try to use viruses as a starting point—they evolved specifically to deliver genetic material into cells.
The primary viral vectors in use are adeno-associated viruses (AAVs) and lentiviruses. AAVs are comprised of a tiny sheath of proteins that self-assemble in an icosahedral geometry around a single-stranded DNA genome with roughly 4,800 bases. The lentivirus family—which includes the HIV virus—consists of larger enveloped viruses that directly integrate their genetic payload into the genome of the host, making them a valuable tool for cell therapies like CAR-T cells.
While there are now hundreds of trials using these vectors, they face serious drawbacks. Viruses are one of the threats that our immune system is the most primed to destroy, which limits their efficacy and poses serious toxicity risks. We’re also limited in the size and type of messages we can store inside of viruses, and in the specificity with which we can target cells. While companies are making progress designing better vectors and scaling our manufacturing infrastructure, developing alternatives to viruses is an appealing angle—especially for heavily engineered RNA payloads.
The catchall of “non-viral” primarily boils down to lipid and polymer-based vehicles. Chemists and nanoscientists have designed polymer systems that form nanoparticle cages. Still, the only FDA-approved uses of these systems have been for targeted delivery of small-molecule payloads. The most widely used system for delivering RNA has been synthetic lipid nanoparticles (LNPs), which form a shell around their cargo. LNPs were the vehicle for both the Moderna and the Pfizer mRNA vaccines.
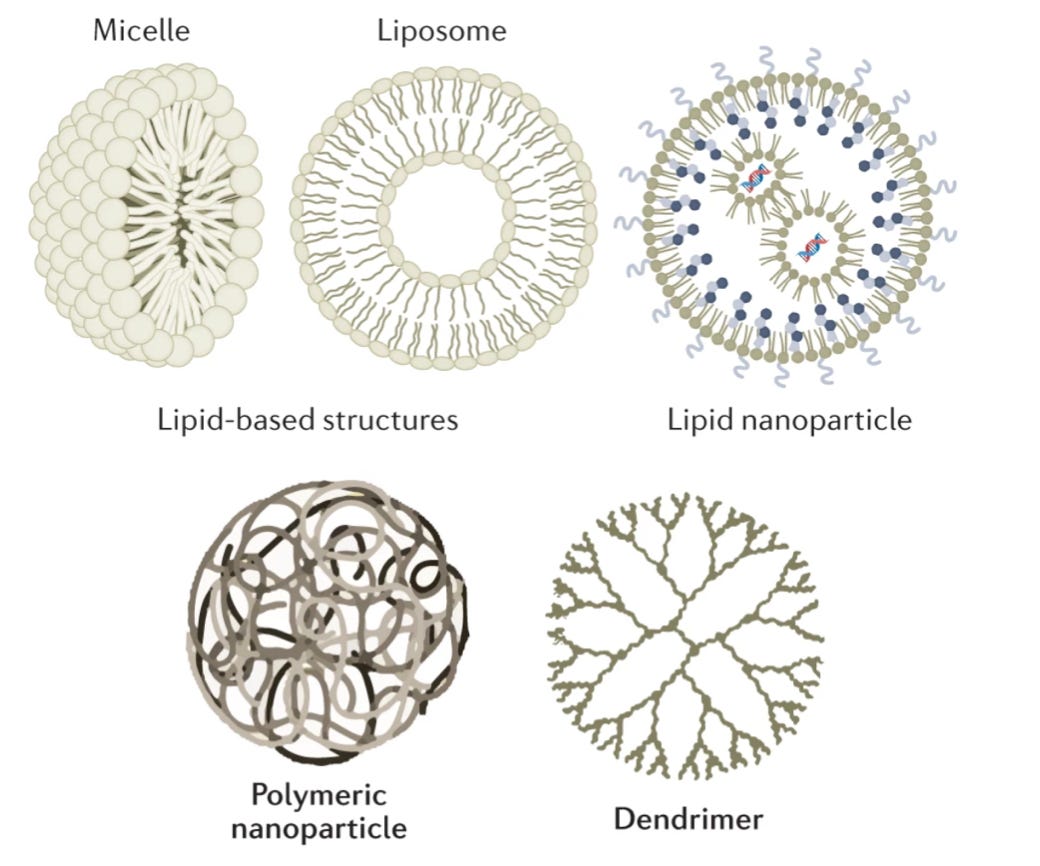
Again, these vehicles are highly imperfect. LNPs notoriously get soaked up by the liver, which evolved to filter out these types of particles. We’re talking about less than 5% of the particles reaching the targeted disease tissue. While developing new approaches for liver detargeting is an active area of research, this shortcoming has skewed early gene therapy trials towards liver diseases—much like how the delivery problem led to a greater focus on retinal disease.
One promising approach to address these shortcomings is to use extracellular vesicles (EVs), which can function as the cell’s own shuttle system for a wide variety of cargo—including nucleic acids.
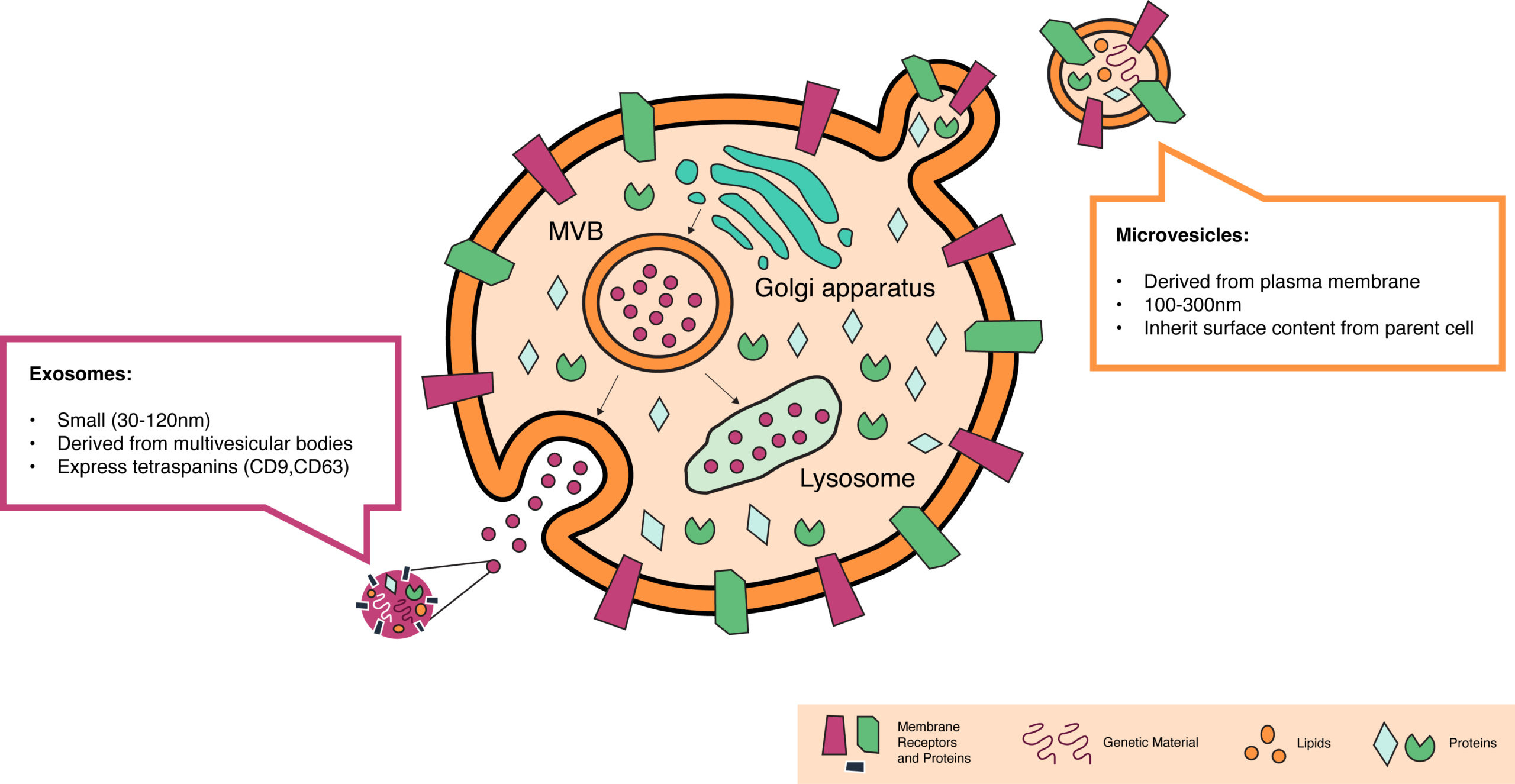
EV biology is directly related to the endocytic-exocytic cycle. We’ve known for a long time that cells have evolved specific machinery to ingest materials from their environment and release some of their inner contents. The ingestion process is called endocytosis. Some cells constantly eat things in their environment—macrophages eat 25% of their own volume of fluid every hour. While this rate is extreme, all cells are continually taking small sips of their environment by pulling in small portions of their own membrane.
Despite this ingestion, cells maintain a constant volume and membrane size because they continually release materials via exocytosis. Cells internally form membrane-bound payloads that are transported to the cell membrane, where their cargo is either incorporated into the cell surface or secreted into the environment.
Our understanding of EV formation within this cycle is much more nascent. We know that when cells undergo apoptosis—programmed cell death—little membrane-bound particles bleb off of the surface of the cell. These particles typically end up being degraded, essentially recycling the cargo. It’s also been documented that cancer cells release small vesicles called oncosomes.

For this reason, extracellular vesicles have mainly been viewed as a sort of garbage disposal for cells. More recently, it’s become clear that EVs have a role as a distinct channel for cell-cell communication. One critical inflection point along this journey was the discovery that exosomes could transfer functional RNA molecules between cells.
Let’s recap some of the biology here and consider why EVs might be valuable as a therapeutic delivery vector—or potentially as a therapeutic on their own.
Just like LNPs, EVs are membrane-bound particles. Exosomes are formed within multivesicular bodies in the endocytic-exocytic cycle (see the first EV figure) and released from the cell. Microvesicles form by directly pinching off of the cell membrane. Both membranes can have a rich set of components that help with immune tolerance and tissue targeting—especially if you’re engineering the cells making the vesicles. It’s been shown that EVs can shuttle various cargo between cells, including proteins, lipids, and functional nucleic acids.
This is the general foundation for the current translational efforts around EVs. The initial efforts have come in a few different flavors. Some early clinical trials have tested using exosomes derived from stem cells as a therapeutic without any specific cargo. The idea here is that these vesicles naturally have membranes and cargo that can improve disease on their own without additional engineering. Now, we’re seeing the first wave of trials using EVs as therapeutic delivery systems.1
One of the major efforts to build an exosome engineering platform was Codiak BioSciences. Codiak was founded in 2015 as a merger between two separate venture creation efforts by ARCH Venture Partners and Flagship Pioneering—two of the biggest players in biotech company creation. The ambitious vision was to dramatically improve exosome therapeutics along three major axes:
Precision engineering — developing systems to incorporate multiple molecular payloads into exosomes.
Manufacturing — building a scalable system for producing exosome medicines for a large number of patients.
Targeting — engineering exosomes to display surface molecules that enhance their delivery to specific disease tissues.
Building on top of this platform, they established a clinical pipeline starting with two engineered exosome therapies. Over time, they aimed to “expand the value of several established drug modalities, such as nucleic acid therapeutics, including ASO, siRNA, miRNA, mRNA, gene therapy and gene editing, to engage targets in a broader range of tissues and cells than the limited set of cells and tissues that currently can effectively be targeted.”
Unfortunately, this won’t be happening.
Codiak filed for bankruptcy last month and plans to find a buyer for its existing assets. In retrospect, this is a cautionary tale for over-capitalizing a company and going public too early. The company cited its serious “financial needs” given that it had $51.8 million in cash, equivalents, and securities, but posted a $19.3 million loss for the previous quarter.2
The silver lining is that this creates a massive opportunity for ambitious scientists and engineers.
EVs are an extremely promising naturally occurring shuttle system for molecular cargo. They have the potential to serve as a sort of universal delivery tool for nucleic acid therapies and could even improve the targeted delivery of small molecules. So far, nobody has been able to make this a reality.
What needs to happen for EVs to succeed? To start, there are still several fundamental engineering challenges to overcome. Just as LNPs have to overcome being soaked up by the liver, EVs have their own set of barriers. After ingestion, endosomes primarily fuse with lysosomes, where their contents are chewed up and degraded. While it’s been demonstrated that EV cargo—including mRNAs—can arrive inside cells functionally intact, degradation will likely still be a bottleneck to overcome for therapeutic effectiveness.
Another direction could be to give more attention to microvesicles—the EVs that are directly released from the cell's outer membrane. STRM.BIO and other companies are pushing in this direction. The proposed advantage is that “microvesicles derived from different cell types have different cell tropisms, making them a highly tunable option for diverse gene therapy applications.”
Whether using exosomes or microvesicles, better measurement technology is critical for the entire field. A recent review on this space stated this clearly: “Simply put, we do not yet have the toolset we need to properly study EVs.” Perhaps rather than directly investing in clinical products and manufacturing as Codiak did, building a platform more squarely focused on better measurement and engineering would be worthwhile. Where is the Dyno for EVs?
There’s been a flurry of new research results in this direction. A team from the Karolinska Institute and Evox Therapeutics posted a pair of preprints describing systems for improved loading and delivery of both protein and mRNA payloads from EVs. A group of synthetic biology researchers at Northwestern also teamed up with David Baker to explore the hypothesis that “designing proteins which associate with ordered, lipid raft-like membranes should increase loading of EVs with engineered protein cargo.” Using the tools of modern protein engineering and the 4-S stack to design enhanced EVs seems promising.
Let’s zoom back out. We’re learning how to program cells but are still struggling to deliver our programs. If we want to reliably cure more diseases using these tools, we need to solve this problem.
Just like we’ve learned to harness CRIPSR for gene-editing and hotwire T-cells to kill cancer, perhaps our solution will come from nature’s own toolbox. EV engineering is an attempt to use an evolved cell-to-cell shuttle system, and I’m excited to see what this field can accomplish.
Thanks for reading this essay about drug delivery using extracellular vesicles. If you don’t want to miss upcoming essays, you should consider subscribing for free to have them delivered to your inbox:
Until next time! 🧬
I’m not sure what to make of this, but a study used plant exosomes to deliver curcumin—the yellow pigment of turmeric—to colon cancer cells.
If you’re here reading the footnotes of this section, you should seriously check out the Codiak episode of the KdT podcast called The World In A Grain Of Sand. In this episode, KdT partners Mack Healy and Rima Chakrabarti did an in-depth breakdown of the Codiak S-1 filing. It provides a perfect foreshadowing of the troubles Codiak ultimately ran into. This is my favorite biotech podcast. Give it a listen!
Add to the list of shuttle systems is the Nanostructured Lipid carrier system (NLC). In contrast to Moderna and Pfizer's Lipid Nanoparticle System (LNP), the RNA here is attached outside the lipid system. By this, the RNA can be replaced much more easily in case of mutation and needs no deep freezing. ImmunityBio does have an ongoing clinical trial, and the system is developed in collaboration with the Advanced Health Institute.