
Safer gene editing
Accounting for human genetic diversity when searching for off-target potential

Armed with the complete CRISPR toolkit, scientists can now exert nearly complete control over both the composition of the genome and its output.
- Jennifer A. Doudna
Overview
Few discoveries have experienced the same escape velocity from the lab to public conversation that CRISPR has had. Since the original landmark paper in 2012, scientists Jennifer A. Doudna and Emmanuelle Charpentier were awarded the 2020 Nobel Prize in Chemistry for their incredible discovery of a programmable method for genome editing.
In addition to the rapid elevation to Nobel status in the scientific community, CRISPR has become a widespread topic of interest and conversation because of the profound ramifications that the system poses for the future of humanity. Having originally evolved as a bacterial defense system to evade viruses, it is now possible for genome scientists to wield CRISPR to make arbitrary genomic edits.
It is hard to overstate the importance of this tool, and the discussions exploring the consequences of its uses have only begun. Two amazing resources for reading more about the discovery, the biology, and the potential power of CRISPR are Jennifer Doudna’s book A Crack in Creation: Gene Editing and the Unthinkable Power to Control Evolution, and the brilliant biographer Walter Isaacson’s new book The Code Breaker: Jennifer Doudna, Gene Editing, and the Future of the Human Race.
One of the most immediately considered (and controversial) applications of CRISPR has been to use it for human gene editing. An obvious use of gene editing is to attempt to use it to cure genetic diseases. While this application is less fraught with ethical and philosophical dilemmas than proposals such as germ-line editing1, it still comes with considerable challenges.
A crucial question to ask is what to edit. This can be enormously complex given the structure of the human genome. Take, for instance the omnigenic model of complex traits, where “most heritability can be explained by effects on genes outside core pathways.” What does that mean? That the majority of the genetic basis for complex traits doesn’t come from the core genes that are relevant to the actual trait.
This idea can be a bit tricky to grok at first, but in a way it is an engineer’s nightmare, making it quite tough to reason about the impact of a given edit and the downstream processes it might be involved in, given the lack of modularity.
In other cases, there are therapeutic targets for gene editing with a less daunting genetic architecture than that of complex traits. Even when the target is more tangible, another technical challenge arises: the potential of off-target edits in the genome.
In order to mitigate the risk of unintentionally editing sites in the genome other than the intended target, considerable effort has been put into developing tools and technologies to better predict off-target potential for designed guide RNAs (gRNA). One shortcoming of current tools is that they are often based on the reference genome2, and fail to account for the possible affects of the natural genetic diversity present in human populations.
To improve upon this, the Pinello Lab at Massachusetts General Hospital and the Bauer Lab at Boston Children’s Hospital teamed up for a new study entitled “Human genetic diversity modifies therapeutic gene editing off-target potential”, where they introduce a new web application called CRISPRme. This new tool incorporates human genetic variation into its search for CRISPR off-target sites, with the goal of enabling safer gene editing.
Key Advances
Prior to this work, there have been several studies that have shown that the genetic variation inherent to all of our genomes can make it more difficult to decisively detect potential off-target sites for CRISPR editing. To illustrate this, I’ll share some of the gold from the gold mine of Science Twitter:

As clearly depicted by this emoji diagram, the presence of natural genetic variants can lead to off-target edits that aren’t possible to predict with the information present in the “typical” reference genome that is so widely used for this type of analysis.
To date, the only way to actively incorporate genetic variation into off-target analysis has been to attempt to use the authors’ code to reproduce the results of the previous studies documenting this problem3. Past this, only one bandwidth constrained command-line tool has been described for this task.
To improve this situation, this preprint introduces CRISPRme, a web-based tool for performing variant aware off-target search. The tool provides the following simple interface for user input:
After submitting the molecular details of the designed edit and choosing the reference genome, thresholds, and annotations to use for the analysis, the search begins. Under the hood, CRISPRme builds on top of concepts and data structures introduced CRISPRitz, (another Pinello Lab tool) to perform a fast and efficient search for off-target sites.4
Once the off-target search is complete, the tool generates a variety of reporting formats describing the results, which include useful visualizations highlighting the findings.
Results
This preprint demonstrates an example use of CRISPRme for “a guide RNA (gRNA) targeting the BCL11A erythroid enhancer that has shown therapeutic promise in clinical trials for sickle cell disease (SCD) and β-thalassemia.” In a recent clinical report, the introduction of progenitor and stem cells edited using Cas9 with this gRNA showed really promising restorative results for the treated patients.
Importantly the studies describing the design and use of this gRNA as a therapeutic “did not reveal evidence of off-target editing in treated cells considering off-target sites nominated by bioinformatic analysis of the human reference genome and empiric analysis of in vitro genomic cleavage potential.” With that in mind, let’s take a look at some of the results from the CRISPRme report for this gRNA:
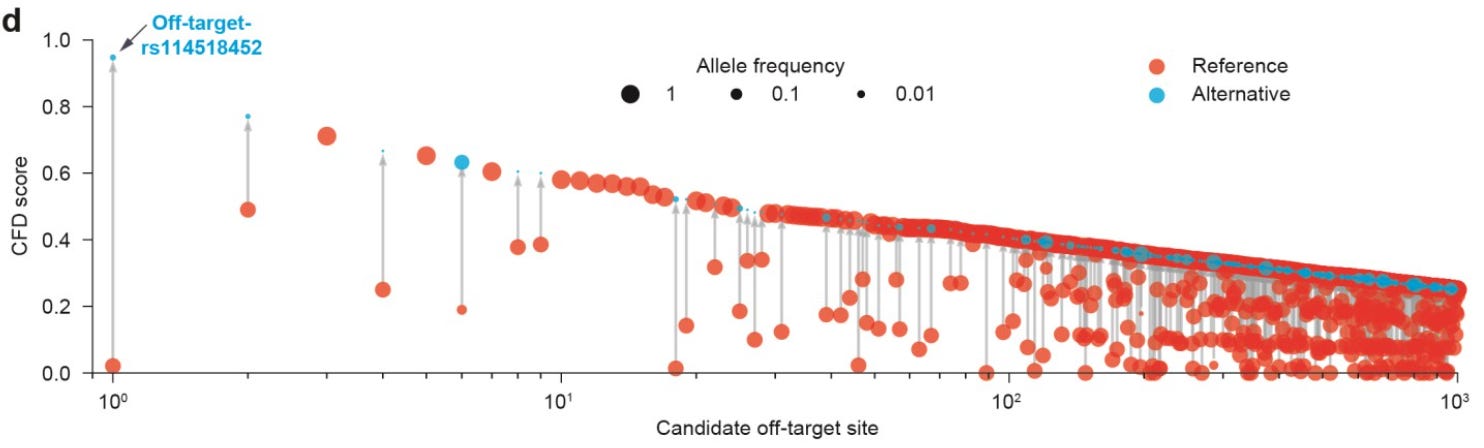
This figure shows the cutting frequency determination (CFD) scores for variants relative to the reference genome, providing a visual depiction of the likelihood of the introduction of a potential off-target site by the variant.
Here, the rs114518452 variant5 is the top predicted potential off-target. In gnomAD, this variant has a non-trivial minor allele frequency in the African population:

This means that this allele could be present in roughly 5% of individuals with African ancestry. The authors designed an experiment to determine whether or not this variant could actually cause off-target effects. They used the gRNA and SpCas9 to edit hematopoietic stem/progenitor cells (HSPCs) with and without rs114518452. They found that “SpCas9 generates indels (~9.6% frequency) and chr2 pericentric inversions in a strictly allele-specific manner” validating this as a legitimate off-target risk that CRISPRme was able to identify.

This represents tangible empirical evidence for CRISPRme’s ability to identify possible off-target sites based on genetic variation.
Final Thoughts
CRISPR represents an incredibly powerful and general tool for making genomic edits. The amount of interest and investment in work to develop CRISPR therapeutics is enormous, and progress is being made at an incredible pace. The ability to edit DNA is a natural complement to the progress that has been made in our ability to read DNA and to write it.
Given advancements on all of these fronts, it is possible to imagine truly futuristic therapeutic protocols becoming a reality. With the steady declines in the cost of sequencing, a patient could have their genome sequenced, or at the very least have their SNPs profiled. This information could be used for personal genome analysis (as demonstrated by tools like CRISPRme) to contemplate and de-risk possible therapeutic edits. At this stage, incorporating the specifics of an individual’s genome, a plan could be devised which would employ an accurate and safe edit, or another type of cell-based therapy.
This type of workflow will rely improvements to our molecular tools (read/write/edit), as well as the continued development of computational methods for solving problems at all of the stages of design and execution.
Thanks for reading this highlight of “Human genetic diversity modifies therapeutic gene editing off-target potential”. If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, have a great Sunday! 🧬
Which would have generational consequences, as opposed to editing somatic cells in people suffering from disease, which would not be heritable genome modifications.
For the people without a genomics background in the audience, research in the field is organized around a “reference genome” of a given organism, such as hg19 or hg38 for humans, or the dm6 reference genome for fly researchers. These serve as a consensus map that facilitates the analysis of sequencing data, and for listing annotations such as genes in a common coordinate system, but we all have bases that differ from these references, making use who we are. There was some big news this week in the world of reference genomes. The Telomere-to-Telemore (T2T) Consortium posted a preprint describing the first complete sequence of a human genome, which will be the study that I will highlight next week. This is a huge milestone in the field of genomics, and highlights the importance of a number of newer sequencing technologies.
This entails a considerable amount of effort and expertise.
In addition to what CRISPRitz can detect, CRISPRme also provides “added support for haplotype-aware off-target enumeration, short indel variants and a flexible number of mismatches and bulges.”