
Resolving the genomic architecture of single cells
Leveraging long-read sequencing to perform high resolution single-cell genomic analysis

Progress in science depends on new techniques, new discoveries and new ideas, probably in that order.
- Sydney Brenner
Overview
It is hard to overstate the importance that methodological advances have played in the history of genetics as a discipline. Even predating the race to deciphering the double helix, genetics has attracted rigorous methodological minds. It is incredible to imagine the scene of the Fly Room, where Thomas Morgan began to systematically investigate the physical basis of heredity by breeding Drosophila melanogaster as early as 1905. The lab was packed with overripe fruit and a perpetual cloud of flies, but Morgan’s focus was on a method that was capable of resolving information deep within cells.
A little bit over a century later, we are capable of routinely generating single-cell genomic data sets that Morgan couldn’t even dream of, but genome scientists around the world continue to push the envelope of what is possible to measure.
I was amazed by the advances being described each week in the lecture hall at the UW Genome Sciences Department where I worked as an engineer. I distinctly remember listening to Junyue Cao’s1 incredible PhD thesis defense. He described a series of new technologies leveraging combinatorial indexing2 to dramatically increase the number of cells that could be profiled in a single-cell sequencing experiment. These tools enabled the creation of comprehensive cell atlases of entire model organisms. By continually advancing what was possible to measure, exciting new biology came into focus. Progress was proceeding as Brenner described.
The entire field of single-cell genomics has been moving at a breakneck speed for many years. However, the technologies in this field build upon short-read Illumina sequencing protocols. While the datasets generated shine light on an enormous amount of new biology, the read length imposes a limit on the type of questions that can be answered. Repetitive regions of the genome can be impossible to correctly resolve from short-read sequencing data, leading to “dark regions” of the genome that remain unstudied. Long-read sequencing can also more effectively identify structural variation, making it possible to connect it to phenotypes.
This week, I’m highlighting exciting new results from the Karolinska Institutet in Sweden, which is continually producing word-class work in genomics. In an exciting preprint entitled “Long-read whole genome analysis of human single cells" led by Joanna Hård from the Michaëlsson and Ameur groups, they describe a new approach for single-cell whole genome sequencing leveraging long reads. This research direction offers exciting new possibilities for answering fundamental questions about the physical architecture of the genome.
Key Advances
At a first glance, it seems technically daunting to consider the prospect of applying long-read sequencing to single cells. At the molecular level, the numbers just don’t seem to add up. For this reason, while single-cell whole genome sequencing has been pursued for nearly a decade, researchers have shied away from using long-read technologies3. The authors highlight some of the technical bottlenecks:
In a diploid cell, only two DNA molecules exist at each locus in the genome, and every molecule that is lost during sample preparation, or fails to be sequenced, inevitably leads to allelic drop-out and missing data. Moreover, the long-read sequencing protocols require large amounts, typically several micrograms, of input DNA. This is about a million times more DNA than what is contained within a single human cell, …
As a starting point, this is a considerable deficit in the raw materials necessary for long-read sequencing. In order to circumvent this bottleneck, some form of DNA amplification is a baseline requirement. However, any type of molecular amplification has the potential to introduce considerable noise and errors into a data set, since even small differences in the number of times a molecule is amplified each round can balloon exponentially into an artifact as the rounds compound.
The key insight in this new protocol was to use droplet-based multiple displacement amplification (dMDA) before sequencing:
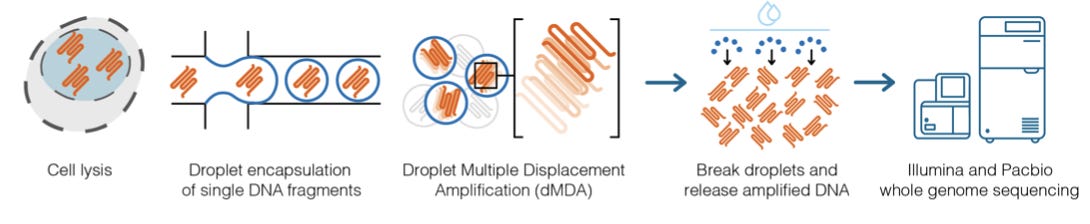
The MDA reaction is pretty interesting:

It has a few advantages over PCR for whole-genome amplification: it uses random hexamer primers, as opposed to sequence-specific primers, leading to amplification of all DNA present, and uses the Φ29 DNA polymerase, which has a lower error rate than Taq polymerase. This leads to an intricately branched network of amplified DNA. Nucleases can then be used to cleave the fragments at the branching points before sequencing.
A key detail that makes this amplification technique particularly amenable to use with long-read sequencing is that the protocol “enables to read molecules of at least 20kb length.” The group was able to establish a protocol that uses dMDA to prepare enough sample DNA for PacBio HiFi sequencing from single cells.
Results
In developing a new measurement technology, it is important to design experiments carefully in order to be able to evaluate accuracy. A large portion of the results in this preprint consist of a careful evaluation of using for dDMA for whole genome amplification. It appears that dMDA provides a more robust and uniform increase in sequencing coverage compared to MDA, with less amplification bias:

This circle plot provides a global visual indication of the increase in coverage uniformity across the genome provided by using dMDA.
While it was heartening to see the confirmation of the amplification technique, I was really excited by some of the examples of the new resolution provided by being able to utilize long-read sequencing:

This type of visual comparison of the reads in a genome browser highlights the power of the long-read sequencing technology. This shows that Illumina reads failed to cover a repetitive region in the CDC73 gene, while this same region was spanned in both PacBio HiFi samples. This is shining light on regions of the genome that were previously left completely unresolved!
There were also promising results about an impressive increase in structural variants detected in the PacBio samples, but I will leave that for curious readers to dig into further.
Final Thoughts
Progress in understanding the genome has been driven by a relentless march of scientists pushing the boundaries of the resolution at which we can measure it. From establishing a culture of model organism communities, to deciphering the double helix, to the modern genome era, each methodological advance shines a light on new biology that we previously couldn’t even detect.
I enjoyed this preprint, not only because it establishes an exciting new proof-of-concept for sequencing single cells with greater resolution, but because it opens the door to exciting future directions. In closing, the authors state
Ultimately, new innovations and technical advances may in the future enable near-complete genome assemblies and full haplotype reconstructions from individual cells. Our work presented here is a first step in that direction.
Thank you for reading this highlight of “Long-read whole genome analysis of human single cells".
If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, have a great Sunday!
Junyue defended his thesis in March, 2019, advised by Jay Shendure. He has now started a new lab at Rockefeller.
The sci-RNA-seq paper is absolutely incredible. However, the difference in scale achieved between this paper and the 2 million cell "mouse organogenesis cell atlas" paper is pretty mind-blowing.
As I mentioned in my post describing SPUMONI, long-read technologies are newer and have made considerable improvements recently in both chemistry and software that are leading to higher adoption.