


Learning the language of variants
Protein language models for genome-wide variant prediction

Welcome to The Century of Biology! Each week, I post a highlight of a cutting-edge bioRxiv preprint, an essay about biotech, or an analysis of an exciting new startup. You can subscribe for free to have the next post delivered to your inbox:
If you want to invest in some of the top companies on the frontier of biology, you should consider joining the Not Boring Syndicate. Make sure to mention The Century of Biology in the application note.
Enjoy! 🧬

Unless you’ve been living under a rock, you’ve probably noticed that AI is starting to get pretty good. Machine learning models have leapt out of the world of games—where they had started to beat humans pretty much everywhere—and become startlingly good at a variety of other tasks. AI models are learning to generate text, code, and even art.

This progress is also spilling over into the world of atoms. One of the areas of natural science that’s been the most rapidly accelerated by AI is protein science. Proteins are molecular machines that are encoded by genes. They are comprised of linear chains of amino acids that fold into a 3D structure. One of the deepest relationships in molecular biology is between structure and function. The 3D structure of a protein dictates its functional behavior when interacting with other molecules.

{kind=link}
Protein structure prediction is the challenge of correctly guessing the final 3D structure for a sequence of amino acids. It’s been an open problem in biochemistry for decades—until recently. In 2020, Google DeepMind produced such an accurate model for this problem that the committee measuring progress in the field announced that “an artificial intelligence (AI) solution to the challenge has been found.”
The sociological response to this result has been fascinating. One talented scientist in the field wrote a famous blog post after DeepMind’s first entry into structure prediction in 2018, asking: “What just happened?” Many have bristled at the excitement and hype, and reminded us all that this one breakthrough won’t revolutionize drug discovery—which is definitely true! But if we zoom out, it’s important to recognize that this is one of the most impactful natural science problems ever solved by AI.1
It’s also important to realize that this is only the tip of the iceberg. DeepMind’s AlphaFold model is the most substantial example of the broader trend of protein science rapidly becoming a quantitative and predictive discipline. There are other important problems, such as learning how to predict the impact of coding variants.
Genetic variants are locations in the genome where bases of DNA vary between individuals. Variants can be placed into two buckets: coding variants and non-coding variants. Most variants are non-coding, meaning that they don’t fall inside of a region that actually encodes a protein. This makes disease geneticists lives harder, because it is even more difficult to mechanistically understand how non-coding variants actually lead to variation in traits.2
Even for coding variants, it’s a non-trivial problem to understand how a given mutation will actually impact a protein. AlphaFold represents a solution to the problem of going from sequence to structure. Predicting the impact of a variant is a question in going from structure to function.
There are two ways to try to understand what a variant does:
Experiment: mutate a protein and actually see what happens.
Computation: use a model to predict the impact the variant will have.
While experiments will always be the gold standard, the number of possible mutations is enormous. Only 2% of missense variants—mutations leading to an amino acid change—have actual clinical interpretations. This leaves a massive number of Variants of Uncertain Significance (VUS) to deal with, which is one aspect of the difficulty associated with scaling the clinical utility of genomics.3
Because of the difficulty of scaling experimentation, computational variant prediction has been an active field of research. Similar to the protein structure prediction problem before AlphaFold, the field has primarily relied on using physical modeling and information that can be gleaned from evolutionary relationships.
In 2021, we saw some of the first major advances for ML-based solutions to this problem. In a landmark paper, the Marks Lab at Harvard developed an unsupervised learning approach to variant prediction. The model was built on top of evolutionary information about the distribution of protein sequence variation across the tree of life. In another testament to the excitement around ML for protein science, Facebook AI Research also entered the field.
The Facebook team expanded on an interesting alternative approach to using information from evolution: training language models on protein sequences. Recent AI breakthroughs in language modeling and generation like GPT-3 have used the Transformer neural network architecture. Although the language of proteins is clearly different from natural language, the logic was that the same attention mechanism in the architecture could potentially learn important aspects of protein structure:
The Transformer processes inputs through a series of blocks that alternate self-attention with feed-forward connections. Self-attention allows the network to build up complex representations that incorporate context from across the sequence. Since self-attention explicitly constructs pairwise interactions between all positions in the sequence, the Transformer architecture directly represents residue–residue interactions.
Admittedly, this isn’t the easiest concept to understand. This architecture was introduced in the foundational paper Attention Is All You Need by the Google AI team, which has now been cited over 50,000 times since 2017. The key idea is that a model can be built entirely around the concept of attention, which is a step in the model that learns a weight for each position in the input sequence to represent its relative significance.
Self-attention extends this by also “relating different positions of a single sequence in order to compute a representation of the sequence.” This is important for modeling language, where it’s necessary to build up an understanding of the relationship between different parts of a phrase or sentence. In the context of proteins, this has now been shown to be a powerful approach for modeling relationships and interactions between even distant parts of an amino acid sequence.
It’s worth noting that a big part of the success we’ve seen in applying ML to proteins isn’t just about a fancy new architecture: these models require massive amounts of data. Just like the natural language models that are trained on an enormous data set generated from crawling most of the Web and parsing a huge number of books, these protein language models were trained on data sets with up to 86 billion amino acids. Between the scale of protein data available and the use of the Transformer architecture, these models achieved state-of-the-art results for predicting the effects of mutations.
Now, in a new preprint from UCSF, researchers have scaled this model to make predictions for all ~450 million possible missense variants in the human genome.
This work was led by Nadav Brandes, and was a collaboration between the labs of Jimmie Ye and Vasilis Ntranos. The core model was the same one that was released by the Facebook team:

The Facebook team had trained on the entire UniProt database of 250 million proteins, and optimized based on the masked language modeling task. Input sequences had several amino acids randomly masked, and the training objective was to correctly predict the amino acid present in these positions. The resulting model had 650 million parameters—which is absolutely enormous—making it expensive to train. Thankfully, the pre-trained model was shared on Github, which served as a starting point for this new study from UCSF.
With the trained model in hand, the authors engineered a solution to circumvent the arbitrary input length of 1,022 amino acids for the model—which would exclude 12% of the proteins present in the UniProt database. Once the model could support protein inputs of all lengths, they thoroughly evaluated how well the protein language model actually performs.
The model is very effective. It slightly outperformed EVE (the evolutionary ML model introduced earlier) on classifying variants from several of the major variant databases. It also performed better on correctly predicting experimental measurements. This performance represents the new state-of-the-art for variant prediction, which is why the authors argue that “protein language models such as ESM1b currently seem to be one of the most promising approaches to determine the clinical and biological consequences of genetic variants.”
With such impressive performance, it’s interesting to look at how the model scored the Variants of Uncertain Significance, which pose a serious clinical challenge:
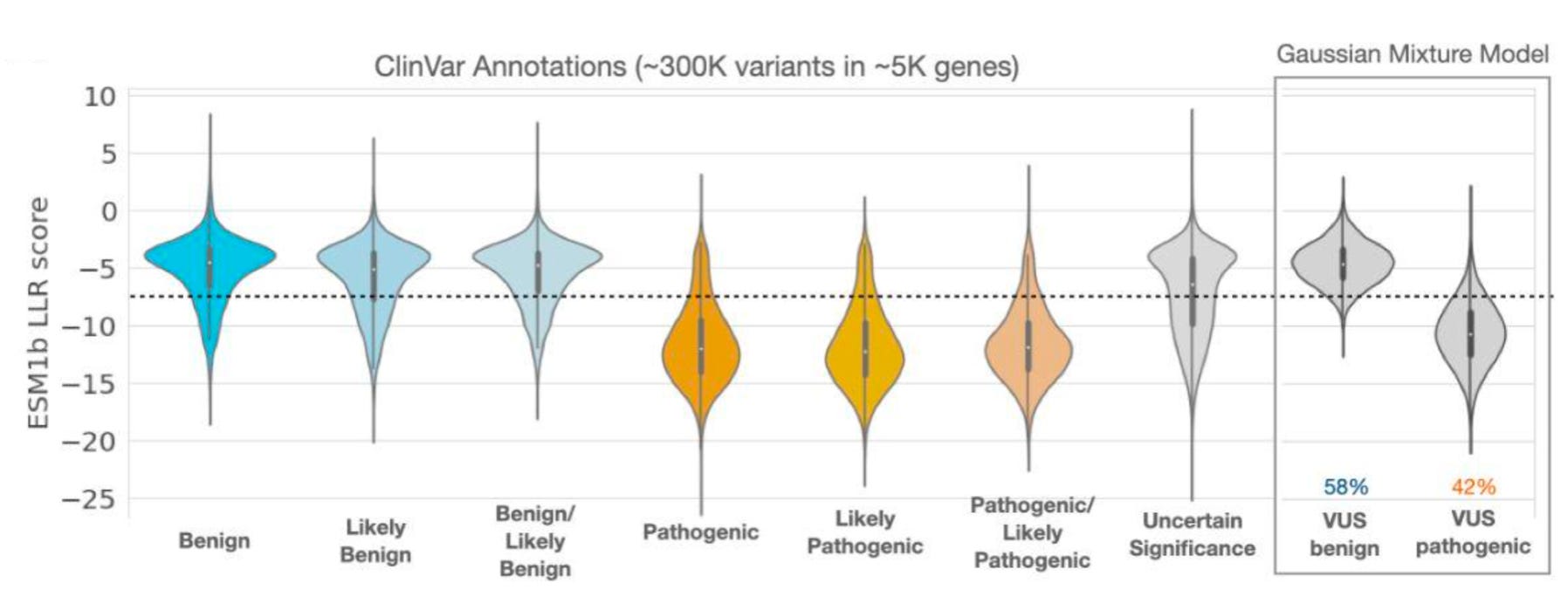
Slightly more than half of these variants were predicted to ultimately be benign. While this is challenging to fully interpret, it’s an interesting insight, especially given the model’s performance on other prediction tasks. It also provides a small glimpse into the future of genomic medicine: how will we handle situations where we don’t fully understand how an AI model scored a variant, even if it is likely to be correct?
We do have some tools at our disposal when trying to interpret how a variant is actually disrupting biological function. One of the interesting results in the paper was that there were thousands of variants with large differences in functional predictions across protein isoforms. Isoforms are slight variations of a single protein, which are the result of processes such as alternative splicing:

{kind=link}
The authors drilled down into a few of the examples in an attempt to interpret the major differences in predictions. Why do some variants disrupt certain isoforms and not others? One of the examples was in MEN1, a tumor suppressor gene. The second isoform of the protein is missing 5 amino acids. Using the model, the authors predict that this small difference creates a region of 30 amino acids that are much more sensitive to variants. It turns out that a large number of studies had already reported missense variants in that region to be associated with cancer, but hadn’t yet explained the functional mechanism. When using AlphaFold—the DeepMind model introduced earlier—the authors noticed that there was a small pocket of the protein which was predicted to be modified by the missing 5 amino acids.

So it turns out that we can also use these models for high impact molecular detective work! It’s likely that there are a number of other interesting lessons to be learned from studying these functional predictions. Thankfully, the authors have released a Web interface to these predictions using Huggingface. Another testament to the power of Open Science!
While this was an exciting step forward for scaling protein language models, there are still many exciting directions open for exploration. For example, how can we more effectively model multiple mutations and the interactions between them?
From an engineering perspective, the potential is enormous. Just this past week, the Baker Lab published a pair of papers (here and here)4 in Science Magazine applying Transformer-based protein models to protein design. These models have been said to be “transforming the field of biomolecular structure prediction and design.” In similar fashion to the original Facebook paper, these models have been made freely available on Github here.
Let’s zoom out and think about all of this in the context of The Century of Biology at large. With genome sequencing rapidly approaching $100 per genome, most people will be able to access it as a service. Tools such as the protein language models discussed today coupled with new large-scale experimental data may dramatically reduce the number of Variants of Uncertain Significance, and potentially even reveal precise information at the isoform level. The therapies prescribed could ultimately be new molecular machines also developed using this powerful new class of models.
Needless to say, the intersection of AI and protein science is worth watching closely.
Thanks for reading this highlight of the use of protein language models for variant prediction. Each week, I post a highlight of a cutting-edge bioRxiv preprint, an essay about biotech, or an analysis of an exciting new startup. You can subscribe for free to have the next post delivered to your inbox:
If you want to invest in some of the top companies on the frontier of biology, you should consider joining the Not Boring Syndicate. Make sure to mention the Century of Biology in the application note.
Until next time! 🧬
Just a few days ago, Demis Hassabis and John Jumper were awarded the Breakthrough Prize for their work on AlphaFold.
There are some extremely impressive efforts to scale experimental variant interpretation at the Brotman Baty Institute in Seattle, as well as in other labs.