
Integrating public data
A new data science method for mapping the relationship between gene modules and disease physiology

The number of people that deposit data greatly exceeds the number of people that withdraw data. It's kind of like a bank, where everyone is taught how to deposit money. No one's been taught how to withdraw money.
- Atul Butte
Overview
For many years, the biomedical data scientist Atul Butte has attempted to convey the stark asymmetry between the production of publicly available biomedical data and its actual consumption. In the culture of genomics and biomedicine at large, we are infatuated with the act of measurement. The genomics revolution is often characterized by the mind blowing cost decline of generating sequencing data. We miniaturize, multiplex, scale, and commodify our data generation technologies. But what are we doing with the mountain of data that we continue to accumulate?
In biomedicine, the concept of secondary analysis of data has not only been under-explored, but at times has been actively discouraged:
A second concern held by some is that a new class of research person will emerge — people who had nothing to do with the design and execution of the study but use another group’s data for their own ends, possibly stealing from the research productivity planned by the data gatherers, or even use the data to try to disprove what the original investigators had posited. There is concern among some front-line researchers that the system will be taken over by what some researchers have characterized as “research parasites.”
Thankfully, instead of shying away, some researchers have leaned into this disagreement, morphing this insult into a complement. A group of computational biologists and data scientists founded the Research Parasite Award, which honors “outstanding contributions to the rigorous secondary analysis of data. This practice of secondary analysis plays a key role in the scientific ecosystem: conclusions that persist through substantial reanalysis are expected to be more credible; and analyses that extract more knowledge from underutilized data make the practice of science more efficient.”
One of the young professors who has staked his career on the idea that there is much to be discovered in public biomedical data is Casey Greene. In describing his lab, he writes “There is an abundance of publicly available data about various biological systems, but it can be difficult to draw insight from individual datasets. Our lab develops algorithms that integrate these data to help model and understand complex biological systems.”
One of the most promising goals of this field is to leverage the large amount of publicly available gene expression data to detect and prioritize potential mechanisms of disease biology that warrant further investigation. In an exciting new collaborative preprint from the Greene Lab entitled “Projecting genetic associations through gene expression patterns highlights disease etiology and drug mechanisms”1 a team of researchers has proposed a clever new data science technique for making progress on this problem. This project was led by Milton Pividori and had contributions from many labs across more than 5 different research institutions!
This preprint certainly lives up to the tweet that sparked the idea for the Research Parasite Award:
Key Advances
While every cell in our bodies contains a copy of the same genome, gene expression patterns between cell types are vastly different. This is the result of complex gene regulatory processes, which control these patterns with exquisite sensitivity. Understanding how the genome is regulated is one of the foundational challenges of the life sciences in the 21st century, and sits at the core of efforts such as ENCODE.
When disease occurs, it is often the result of a deviation from tissue-specific regulatory processes. What this means is that “determining how genes influence these complex phenotypes requires mechanistically understanding expression regulation across different cell types.”
How can we decipher these types of mechanisms? One methodological approach is the Transcriptome-Wide Association Study (TWAS). Backing up a bit, a Genome-Wide Association Study (GWAS) is a widely used experimental paradigm for detecting genetic variants associated with a trait.

{kind=link}
On the other hand, a TWAS “integrates expression quantitative trait loci (eQTLs)2 data to provide a mechanistic interpretation for GWAS findings.” Okay, so we are trying to explain genetic associations at the level in which they are directly impacting molecular mechanisms by impacting gene expression. Conceptually, this is a great step forward!
However, there is a problem where the rubber meets the road: “TWAS have not reliably detected tissue-specific effects because eQTLs are commonly shared across tissues.” Given the fact that eQTLs are shared across tissues, you can’t purely rely on TWAS alone to decipher the way in which tissue expression goes awry in the disease state based on genetic variation.
This is where the approach called PhenoPLIER presented in this preprint comes into play:
We integrated more than 4,000 gene-trait associations (using TWAS from PhenomeXcan) and transcriptional proles of drugs (LINCS L1000) into a low-dimensional space learned from public gene expression data on tens of thousands of RNA-seq samples (recount2). We used a latent representation defined by a computational approach that learns recurrent gene co-expression patterns with certain sparsity constraints and preferences for those that align with prior knowledge (pathways). This low-dimensional space comprised features representing groups of genes (gene modules) with coordinated expression across different tissues and cell types.
This sounds like a lot to unpack, but at its core there is a really clever conceptual simplicity. This can be seen when the workflow is represented visually:

At the top of the diagram, we see that a tool called MultiPLIER is used to factor out what is called a latent gene expression representation from the recount2 database. Importantly, “Each of the 987 latent variables (LV) represents a gene module, essentially a group of genes with coordinated expression patterns (i.e., expressed together in the same tissues and cell types as a functional unit).”
Additionally, we see that a large set of GWAS results was integrated with eQTL data using PhenomeXcan to generate a matrix of associations between genes and traits. Finally, this matrix is combined with the latent representation from MultiPLIER to produce a matrix of traits associated with the latent variables (gene modules).
Using a carefully architected data integration strategy, this project was able to arrive at a novel data representation that could be used to study the relationship between traits and how genetic variation affects functional gene modules at the level of tissues. This is some really cool data science!
Results
One initial question to investigate is how much new signal these module-based associations provide relative to previous gene-based associations. This preprint evaluated this by measuring how well these two approaches “accurately predicted known treatment-disease pairs.”
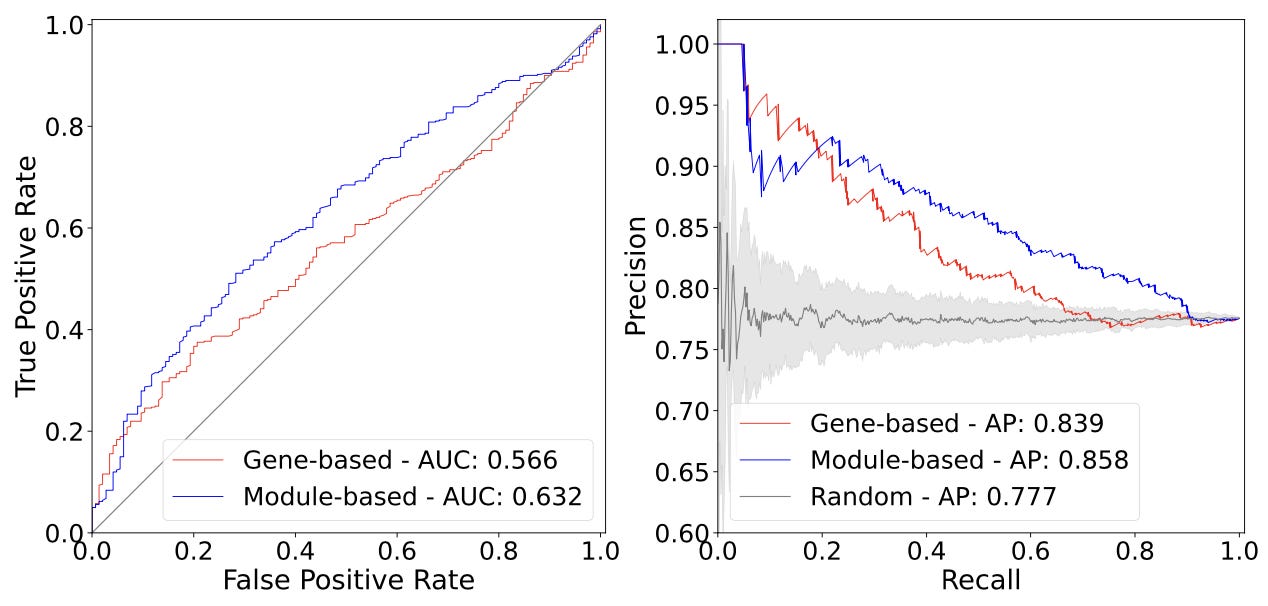
From the ROC and PR curves, you can see that there is a gain in predictive performance in using gene modules compared to making predictions directly based on genes. While these numbers aren’t enormous, data integration across many studies and resources is a noisy process, and this seems like an interesting result that indicates that this new method is working as expected. In data mining, even this type of gap between curves represents the possibility to uncover a lot of new interesting biological associations!3
There are a number of cool things to do with this new representation that links traits to gene modules. One of them is to generate new primary data for a phenotype of interest, and use it to steer the exploration and validation of this new representation. In this study, they generated new data to detect genes associated with lipid accumulation using a CRISPR-Cas9 screen. They derived two gene sets: 1) a set of 96 genes causing a lipid decrease, and 2) a set of 175 genes causing a lipid increase.
Next, they mapped this data back onto the LVs (gene modules). They retrieved the LVs enriched for these gene-sets for further inspection. In particular, there were 27 associated with the lipid increase gene set. These LVs were also associated with a “diverse set of traits, such as blood count tests, impedance measures, and bone-densitometry.”

As can be seen above, one of these LVs (LV246) is heavily enriched for genes expressed in adipose tissue, “which plays a key role in coordinating and regulating lipid metabolism.”
There was an interesting result to tease apart here in evaluating whether or not this type of module level association could be made just by evaluating gene-level associations with traits. One interesting patten in this module was that “Two high-confidence genes from our CRISPR screening, DGAT2 and ACACA, are responsible for encoding enzymes for triglycerides and fatty acid synthesis and were among the highest-weighted genes of LV246.” But at the gene association level:
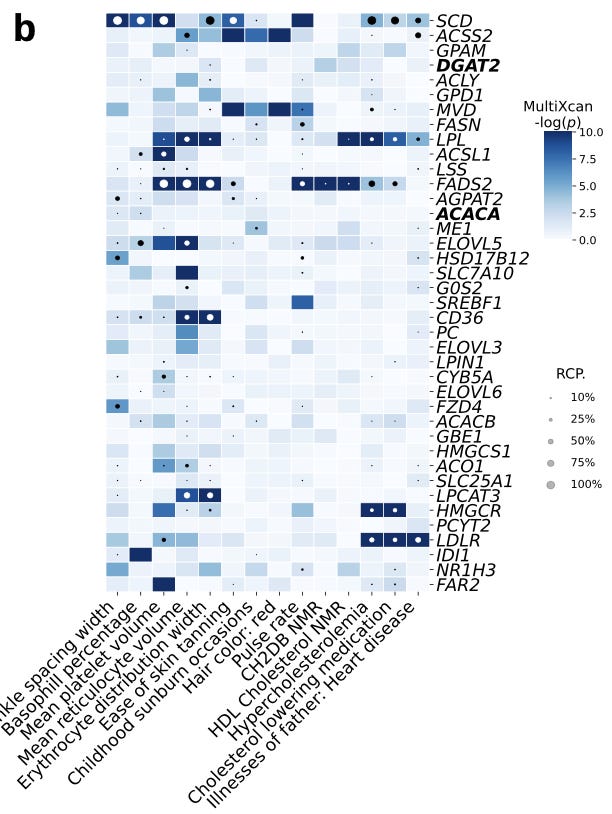
These two genes were not strongly associated with these traits using TWAS alone. This case study for lipid metabolism shows an example of how shared eQTLs across tissues can drown out the signal of underlying mechanistic associations, whereas these types of relationships can be teased out of the latent variables.
Another segment of this study performed cluster analysis to explore this data and the clusters of traits with shared transcriptomic properties. Reading this portion of the paper is a good exercise for those interested in unsupervised machine learning. All sorts of weird and interesting biological associations came out of this analysis. For example: “Within the cardiovascular sub-branch, we found neuropsychiatric and neurodevelopmental disorders such as Alzheimer’s disease, schizophrenia, and attention deficit hyperactivity disorder (ADHD).”
Biology is really complex! This type of finding represents the way in which complex traits can be composed of simpler underlying gene modules that are shared across other vastly different traits. Interestingly, “none of these LVs were aligned to prior pathways, which might represent potentially novel transcriptional processes affecting the cardiovascular and central nervous systems.” In addition to being a new aspect of biology to explore and better understand, this also represents a pragmatic opportunity to repurpose existing drugs that may exist for these different traits that target the same underlying modules.
Final Thoughts
I love new data just as much as the next scientist. However, I sometimes wonder about how many decades worth of new findings simply lurk within the incredible public data resources that we all take for granted. I find it really compelling to see new methods being derived to integrate these large compendia and harness them to uncover underlying biological patterns. This type of much-needed parasitism only stands to make the ecosystem of modern biomedical research more robust and efficient.
Thanks for reading this highlight of “Projecting genetic associations through gene expression patterns highlights disease etiology and drug mechanisms”. If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, have a great Sunday! 🧬
You may be curious about the typesetting and formatting of this paper. It was created using Manubot: a system designed for open and collaborative academic writing using Github and version control. This project was pioneered by Daniel Himmelstein (while a postdoc with Casey Greene) and Anthony Gitter. The system holds some really exciting and inspiring ideas for the future of open scholarly publishing. It enables the creation of interactive, web-based publications that can be continually updated, such as the “Deep Review.”
Expression quantitative trait loci (eQTLs) are genomic loci that explain variation in expression levels of mRNAs.
After reading this post, Casey Greene raised a great point. I wasn’t quite thinking about these results correctly. Here, the gene module representation is actually a compression for the purposes of interpretation and discovery. Typically in machine learning, compression results in slightly worse performance, because it is a loss of information. Given that, it is actually pretty impressive and interesting in the case that it performs as well, let alone better!