
Genotype-phenotype maps
The first genome-scale Perturb-seq experiment

Genetics works.
- David Kingsley
Introduction
Arguably no technology has improved as rapidly as DNA sequencing. It can be useful to benchmark progress against the incredible acceleration in the processing power, miniaturization, and commoditization of computing that we have lived through. This trend has tangibly changed and disrupted all of our lives already.

This widely used graph shows how the decrease in sequencing costs has actually dramatically outstripped even Moore’s Law—the predicted doubling of the number of transistors in a dense integrated circuit (IC) every two years—which has been used to forecast the digital revolution.
It’s hard to overstate how important this change has been for the life sciences. Over the course of the 20th century, the language of genes has transformed nearly every field of biology. As David Kingsley would say, this is because “genetics works.” It provides a unique and central toolbox for describing complex biological phenomena with mechanistic detail. As genetic technology rapidly advanced, the scope of research that was possible dramatically expanded. Only a few decades ago, a cutting-edge PhD thesis at a top institution would describe the sequencing of a single gene. Now, graduate students routinely sequence entire genomes, even across multiple organisms.
In recent years, mechanistic genetics has also changed dramatically. Well before the sequencing revolution and the birth of genomics, one of the primary approaches to mapping and discovering genes was forward genetics. This experimental framework consists of starting with a phenotype—an observable biological trait—and then working to map the underlying gene responsible. The molecular biology revolution made it possible to start doing reverse genetics, where research starts with the alteration of a gene and then works to detect the phenotypic consequences.1
The discovery of CRISPR has been a total game-changer for reverse genetics. It has made it possible to program DNA edits, dramatically decreasing the experimental difficulty associated with altering genes. Outside of edits, the CRISPR toolbox has been extended to activating genes and interfering with genes without directly changing their sequences.
What kind of science becomes possible when we combine the two superpowers of DNA sequencing and CRISPR?
Well, graduate students could potentially perturb every gene in the genome and measure the global transcriptional consequences of each perturbation in a single study. This would blend both forward and reverse genetics, creating a dense catalog of both genetic and phenotypic information. In the new preprint “Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq” this is exactly what happened. This massive 40+ page study with 7 figures was led by UCSF graduate students Joseph M. Replogle and Reuben Saunders. The corresponding authors are Thomas Norman and Jonathan Weissman.
Key Advances
So what is a Perturb-seq? It is a technology that couples single-cell RNA sequencing with pooled genetic perturbations typically done with CRISPR. In other words, a large library of CRISPR guide RNAs targeting many different genes is introduced to a population of cells, and then the cells are sequenced. Figuring out which perturbation is associated with each cell can be done by directly capturing and sequencing the guide RNA in a given droplet:

The screen was done in two cell lines: chronic myeloid leukemia (CML) K562 cells and RPE1 cells, both engineered to express dCas9-KRAB, which is a catalytically inactive version of Cas9 that doesn’t cut DNA, fused with a KRAB domain which represses the gene.2 Ultimately, the dataset consisted of “>2.5 million high-quality cells with a median coverage of >100 cells per perturbation.”
With this enormous data set, they set up a statistical framework to be able to detect the transcriptional phenotypes caused by all of the different perturbations. In their preliminary analysis of changes to transcription, they found that 31.1% of the 9,608 different CRISPR perturbations caused a significant transcriptional phenotype. Similarly, they found that 30.5% of the the perturbations led to >10 genes being differentially expressed.3
It’s worth noting how this setup differs from how Perturb-seq has been used up to this point. While sequencing costs have plummeted, it still hasn’t been practical for researchers to perturb every expressed gene in the genome for a given study. Instead, the experiments would use target panels of genes thought to be involved in specific mechanisms. For example, in the original paper in 2016 describing Perturb-seq, one of the panels used targeted 24 transcription factors. That study sequenced a total of 200,000 cells.
A little over five years later, this experiment involved targeting all expressed genes in two different cell lines, and sequencing millions of cells. This is incredible.
Results
With this enormous catalog of genetic perturbations linked to transcriptional phenotypes, they attempted to use this information to annotate gene function. For this analysis, they focused on roughly 2,000 genes with the most pronounced effect when perturbed. They entered this set of genes into databases for known protein complexes (CORUM) and protein-protein interactions (STRING). The gene pairs that had correlated expression profiles were enriched in both results:

Essentially, the genes with similar expression when perturbed appeared to represent actual protein complexes and protein interactions.
They continued with their gene annotation analysis, and found 64 distinct clusters representing an enormous range of processes in cell biology:

This wasn’t simply an exercise in recapitulating known biology. Within these clusters, there were many genes that didn’t have previously described roles—making it possible to use the function of known genes nearby to better understand what they do. In this study, they decided to follow up on a set of 10 uncharacterized genes from the cluster of ribosomal genes. They did another experiment perturbing these genes with CRISPRi, and measured the impact on ribosome biogenesis using the ratio of 28S to 18S rRNA as their readout.4
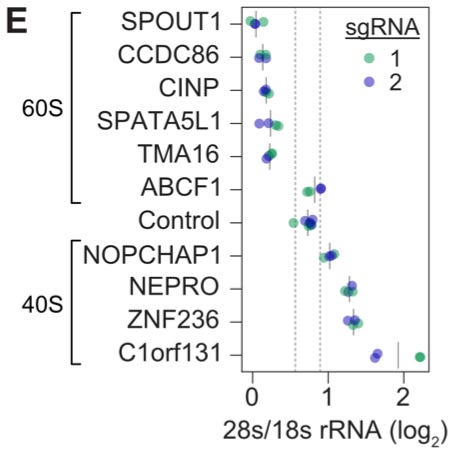
It’s interesting to think about why this type of annotation of unknown gene function was possible in this experiment. The authors make an interesting argument that “while the number of perturbations scales linearly with experimental cost, the number of pairwise comparisons in a screen—and thus its utility for unsupervised classification of gene function—scales quadratically.” Doing this type of screen on the genome scale opens up new directions for analysis.
This analysis attempted to annotate gene function. Another important analysis was done with the goal of characterizing the impact of all the different perturbations. They attempted to answer this challenging question with a careful unsupervised clustering analysis. First, they clustered genes together based on having similar responses across different perturbations—representing potential gene expression programs. Next, they clustered the perturbations together that resulted in similar transcriptional responses. Finally, they looked at the activity of these gene expression programs within the different perturbation clusters.

With this map, they could determine what changes to core gene expression programs were actually being driven by specific perturbations. What are the actual phenotypic consequences of perturbing a given gene? One key question they tried to address was which perturbations led to cellular differentiation. Cellular differentiation is both a fundamental topic of study in developmental biology and a promising therapeutic strategy—differentiation therapy is an active research direction for treating cancer.
One of the current targets of differentiation therapy is the gene KDM1A. In their analysis, they found an additional perturbation that drove differentiation—the inactivation of the PTPN1 gene.

When testing this, both genes drove differentiation, but the strongest response was seen when both genes were targeted.
Final Thoughts
So far, I’ve talked about how the experiment in this study perturbed every expressed gene in the genome in multiple cell lines, annotated the function of unknown genes, and found new targets for differentiation therapy. This could be several standalone studies, but have only scratched the surface of the results in this paper. If you want to dive deeper and explore this paper, they also did several other analyses, including a functional analysis of the Integrator complex, an exploration of composite phenotypes, and deep dive on stress-specific regulation of the mitochondrial genome.
As the sci-fi author William Gibson famously said, “The future is already here. It's just not evenly distributed yet.” He argues that his books don’t bear an uncanny resemblance to the future based on a complex model of technological progress—or even a deep understanding of it. Simply observing what is already happening on the frontier is a powerful way to attempt to understand what will come next.
The enormous experiment in this study is still far too expensive for most scientists to pursue. Despite that, it represents an incredible demonstration of the capacity we are building up in biotechnology—especially at the interface between DNA sequencing and CRISPR. The scope of any single PhD project in biology has dramatically expanded, and we are witnessing a new generation of biotech startups wielding these enormously powerful tools. In the near future, the scale of this Perturb-seq experiment could potentially be commonplace for the majority of biologists and biotech startups. What will biology look like then?
Thanks for reading this highlight of “Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq”. If you’ve enjoyed this post and don’t want to miss the next one, you can sign up to have them automatically delivered to your inbox:
Until next time! 🧬
I’ve heard various arguments about whether or not it would make more sense to flip the definitions of forward and reverse genetics around. I personally think the more confusing naming scheme is introns and exons.
There are two points worth noting here: there are several advantages to using CRISPR inactivation (CRISPRi) here instead of making edits. You can tell how well the inactivation works from the transcriptional readout, it works more consistently, and is less toxic to cells. Also, the RPE1 cells used a different KRAB domain than the original CRISPRi domain based on results from previous work.
There was a correlation between the magnitude of the growth defect in the cell and the transcriptional response. A gene being highly expressed was also predictive of a strong transcriptional response to perturbation.