
Detecting structure in the microbiome
A new computational approach for analyzing microbial communities using metagenomic data

Overview
As descendants of apes, it can be challenging to try to make sense of the universe that we inhabit. Science provides a powerful technical and cultural framework for making progress in gaining understanding, but it can still be challenging to decide what to focus on.
Some people never decide on a topic to focus on. After completing his PhD in biochemistry in 1948 and spending some time in a faculty position, the prolific science fiction author Isaac Asimov “rebelled” against becoming any further of a specialist, saying
I have never been sorry for my stubborn advance toward generalization. To be sure, I can't wander in detail through all the orchard, any more than anyone else can, no matter how stupidly determined I may be to do so. Life is far too short and the mind is far too limited. But I can float over the orchard as in a balloon.
Indeed, the orchard of Science possesses immense variety and beauty. When attempting to find a good location to land, it can be interesting to consider the various motivations for a given field of inquiry.
In biomedical research, there is often considered to be a single axis that spans between “basic” (fundamental) research, and “translational” (directly useful) research. The political scientist Donald Stokes argued that these pursuits are actually two orthogonal axes, meaning that they can be pursued together:

He referred to the top right quadrant as Pasteur’s Quadrant. Louis Pasteur is an example of a brilliant scientist whose work simultaneously advanced our fundamental understanding of the world and was directly applicable to the improvement of the human condition. I first heard about Pasteur’s Quadrant after listening to a wonderful lecture by Stanford physician-scientist Dr. Ami Bhatt. Dr. Bhatt argues that modern microbiome research exists in Pasteur’s Quadrant.
One of the motivating facts for studying the microbiome is that there are somewhere between 1 to 10 times as many bacterial cells in the human body compared to human cells. The enormous range in this estimate for a data point that is typically included in the elevator pitch for microbiome research reflects the tremendous room for increasing our understanding of this system.
In addition to being a frontier for better understanding human physiology, there is an incredible amount of evidence indicating that the microbiome plays a role in a large number of diseases. It appears that microbiome composition has an impact on everything from cancer to mental health (via the gut-brain axis).
It seems like if we can improve our fundamental understanding of the microbiome, we will be poised to dramatically improve human health. This general thesis has spurred a tremendous amount of excitement, research, and company creation.
In similar fashion to the other problems I’ve written about so far in genomics, progress in microbiome research has been largely driven by: 1) better measurement technologies (primarily sequencing), and 2) development of new computational tools for improved inference from data.
One of the pioneers on both fronts has been Dr. Rob Knight from UC San Diego1. The Knight Lab has played a leading role in a number of the largest and most important microbiome projects, and has developed several of the more widely used analytical and experimental tools in the field.
Recently, the Knight Lab published a new preprint entitled “OGUs enable effective, phylogeny-aware analysis of even shallow metagenome community structures” led by Qiyun Zhu and Shi Huang. This work describes a new computational method for microbiome analysis. I am going to highlight why this seems like an exciting new approach.
Key Advances
A key methodological goal in microbiome research is to figure out which organisms are present in a given individual. Knowing this information, it becomes possible to develop more complex inference techniques for understanding how the system might behave collectively, and to discover causal relationships between microbiome composition and various phenotypes in question.
One of the primary tools for develop taxonomies of the microbes present in a sample has been to study 16S rRNA. 16S rRNA encodes a component of the prokaryotic ribosome, and is highly conserved because it plays a critical role in the initiation of protein synthesis. Because of its high degree of conservation, any changes in the sequence can be used to reconstruct the phylogenetic tree of the organisms present.
In contrast to amplifying and sequencing 16S rRNA, shotgun metagenomics sequences the entirety of the DNA that is present, which increases the resolution of what can be studied but introduces new complexity to analysis. In addition to the more complete sequencing data generated, the authors cite a few key recent improvements in genomics and metagenomics: 1) more efficient sequence alignment algorithms, 2) expanded databases of microbial reference genomes, and 3) better phylogenetic trees.
The authors capitalized on these recent advances to develop a new feature to be measured: the Operational Genomic Unit (OGU). The structure of this feature is fairly conceptually simple. An OGU consists of the microbial “individual reference genomes from a database, and the feature counts are the number of sequences aligned to these genomes.”
Prototypes of this data structure have been used in studies by this team before, but this new work establishes a more clear evaluation and introduces a new computational tool for generating OGUs.
Results
The evaluation of OGUs in this paper came in the form of one synthetic example and two case studies of the application the method to existing datasets. The example on a synthetic dataset is useful for building intuition as to why this data structure is advantageous:
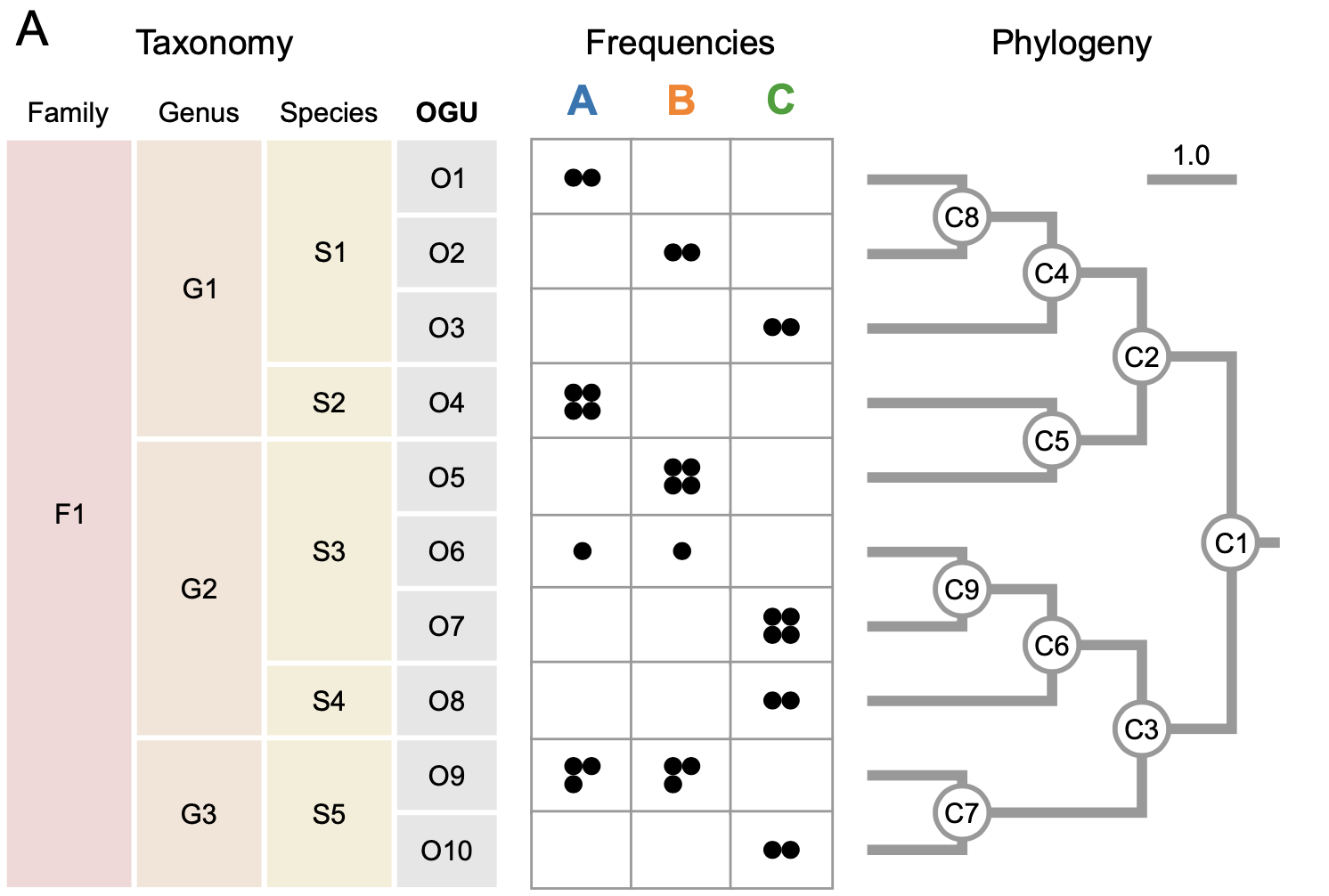
The frequency table is a visual depiction of an OGU. Each column (A, B, and C) represents a hypothetical microbial community. Each row represents the frequencies of read hits to each OGU (again, this is a microbial reference genome in a database). On the right hand side, a phylogenetic tree constructed from the frequency table is shown. On the left hand side, the genomes are grouped taxonomically: first by species, genus, and then family of organism.
Looking at this representation, two things are noticeable. The first is that there is visibly more structure to the phylogenetic tree compared to the taxonomic grouping, but in this simple synthetic dataset, the difference isn’t tremendous. The second aspect is perhaps more interesting: the fifth genome (05) is placed in a different region between the two analysis techniques. This is an example of higher resolution provided by the OGU over taxonomic analysis.
The authors provide a specific definition of this:
This “resolution” has two dimensions of meaning: first, the quantity of features representing individual microbiomes; second, the granularity and accuracy of the hierarchy—if any—that defines the relationships among individual features.
By measuring more information from each sample using shotgun metagenomics it is possible to generate richer genomic data structures. These data structures can be used to build more complex and accurate models of the microbiome.
One exciting result of this study was that OGUs make it possible to detect interesting microbial community structures even in “shallow” sequencing datasets. The team used the Human Microbiome Project (HMP) dataset, “which contains 210 metagenomes sampled from seven body sites of male and female human subjects” and downsampled to a million paired-end reads, to simulate a dataset with shallower sequencing.
This (beautiful and dense) figure provides a visual representation of some of the high resolution community structure detected from the shallow dataset2:

At a high level, the variation in color in the two outer bands labeled “host sex” and “body site” represent differential abundance of the phylogenetic clades. Local community structures in the microbiome vary by sex and body site, and it is still possible to resolve this with shallow read depth using Operational Genomic Units.
While there was also an exciting demonstration of improved accuracy in age prediction from microbiome data with OGUs, the last aspect I’m going to highlight in this post is the computational tool itself. This study introduces Woltka, a Python package for generating OGU tables. The software is freely available on Github.
In academic software, there is a wide range of adoption of software engineering principles. While a considerable emphasis is often placed on novelty, there is often less thought given to ease of installation, documentation, testing, and continuous integration. While developing cutting-edge functionality is important, the latter aspects of software can often be the differentiating factor in whether a new tool is adopted by other researchers and can be maintained or extended with minimal effort over time.
Woltka excels on these fronts. The code is clear and readable, and comes with accessible documentation and a minimal installation process. It also boasts 100% unit test coverage, run in continuous integration on Github. While the percentage itself isn’t the critical part, these tests are high quality infrastructure that will enable the developers to maintain this resource and keep it up to date with minimal effort even when they have moved on to other projects.
On this note, I think that there are lessons to be taken from the field of microbiome research about effectively distributing scientific software for reuse. This tool is also distributed as a QIME 23 plugin. QIME 2 is an extensible software platform for microbiome analysis that has gained fairly broad community adoption. This type of effort for scientists to mix and match various analytic tools for their own projects more easily, and increase the rate of reuse and standardization of methodology.
Woltka represents an advance in the resolution at which we can model the microbiome, and the authors have done the engineering and documentation necessary to make their work readily adoptable by other researchers.
Final Thoughts
I always enjoy reading papers describing microbiome research because it is very often an exciting blend of cutting-edge data science and software engineering, sequencing technology, and really interesting discovery-oriented science. With all of that being said, it is often very tangible how the technology or knowledge produced could be harnessed to reduce human suffering. In other words, it often inhabits Pasteur’s Quadrant. I think that this paper was a beautiful example of this.
There is a tremendous amount still left to discover. Ed Yong describes the microbiome in the following way:
When Orson Welles said “We’re born alone, we live alone, we die alone”, he was mistaken. Even when we are alone, we are never alone. We exist in symbiosis—a wonderful term that refers to different organisms living together. Some animals are colonised by microbes while they are still unfertilized eggs; others pick up their first partners at the moment of birth. We then proceed through our lives in their presence. When we eat, so do they. When we travel, they come along. When we die, they consume us. Every one of us is a zoo in our own right—a colony enclosed within a single body. A multi-species collective. An entire world.
Woltka provides a new tool to better understand that world, and in doing so provides a new way to better understand ourselves.
Thank you for reading this highlight of “OGUs enable effective, phylogeny-aware analysis of even shallow metagenome community structures”.
If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, have a great Sunday!
More background on Rob Knight can also be found in I Contain Multitudes by Ed Yong. This book is an incredible resourcing for learning more about the microbiome from one of the top science writers of our time.
This figure can be a bit difficult to interpret without the full figure caption: “Differentially abundant phylogenetic clades by host sex inferred using PhyloFactor and visualized using EMPress on the WoL reference phylogeny. The tree was subsetted to only include OGUs detected in the dataset. The top 20 clades by effect size are colored (full details provided in Figs. S4-5). The top five clades are numbered 1 through 5 by decreasing effect size, circled, and labeled with corresponding taxonomic annotations. The small color ring represents phylum-level annotations. The inner and outer barplot rings indicate the OGU counts split by body site (using the same color scheme as in A and B) and by host sex, respectively.”
The acronym stands for Quantitative Insights Into Microbial Ecology.