
A new breakthrough for deep learning in genomics
An improved model architecture for incorporating long-rang information into expression prediction from DNA sequences

Overview
This week I am highlighting the new preprint “Effective gene expression prediction from sequence by integrating long-range interactions”, which describes a new deep learning architecture for genomics from Google. The results in this preprint have generated a considerable amount of buzz in the computational genomics community. This paper is also of serious personal interest to me, since I spend a lot of time thinking about the application of machine learning to biology.
In order to cover this paper effectively, I’m going to make an effort to provide a short summary of some of the historical context surrounding this paper that I think makes it especially meaningful. In doing so, I hope that people from a variety of backgrounds can build intuition for what makes the intersection of deep learning and genomics so promising.
The birth of computational genomics
Computation has been essential for the young field of genome biology since its inception. One of the reasons that we have a publicly available genome sequence in the first place is that during the race to its completion, UC Santa Cruz graduate student Jim Kent worked relentlessly to develop GigAssembler. GigAssembler is a software program responsible for assembling the original reference sequence of the human genome from the enormous swirl of sequencing data generated by the Human Genome Project. According to the legendary genome scientist David Haussler, Kent “had to ice his wrists at night because of the fury with which he created this extraordinarily complex piece of code.''1
From the original genome assembly to the birth of genome browsers, bioinformaticians and computational biologists have played a foundational role in the analysis of genomes. As the size and number of genomic datasets has skyrocketed, some of the most crucial bottlenecks in the generation of new knowledge from the data have been computational. In fact, it has even been argued that in the modern era, all biology is computational biology.
The origins of deep learning2
In parallel to the rapid growth of genomics in the life sciences, recent progress in machine learning has revolutionized the field of computer science. For many years, machine learning existed on the fringes of academic artificial intelligence research. The field is structured around the concept that it is possible to learn arbitrarily complicated functions and patterns from data. Theoretical results such as the Universal Approximation Theorem from as early as the 1980s seemed to show that it may be possible for a type of machine learning model called an artificial neural network to learn any function that existed in a data set.
While these ideas were exciting, they only began to produce meaningful empirical results with the advent of cheaper and faster hardware that enabled parallel computing, such as graphics processing units (GPUs), which were originally optimized for video games. Better hardware also enabled “deeper” networks to be trained (models with more layers of artificial neurons), leading to the birth of the term deep learning.
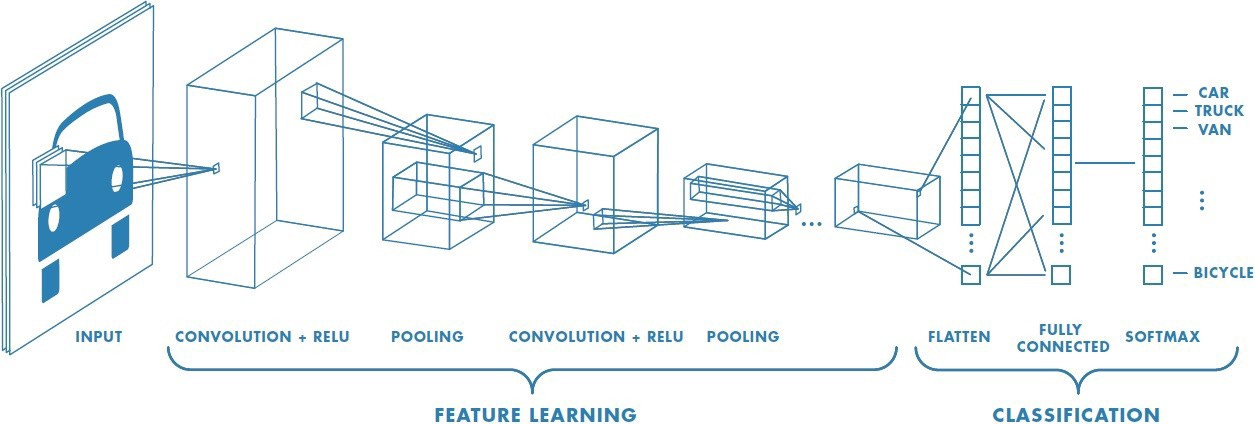
One of the crucial early results in the history of deep learning was a model called AlexNet. AlexNet was one of the early implementations of a convolutional neural network (CNN) using GPU acceleration. A CNN is a specific deep learning architecture that can be used to detect and classify images. AlexNet absolutely dominated one of the central image recognition challenges in AI, winning by a large margin of 10.8% percentage points compared to the second place finisher. This marked a turning point in the adoption of deep learning.
Fast forwarding to the present moment, AlexNet has been cited over 70,000 times, and deep learning has resulted in dramatic progress in several fields of artificial intelligence such as computer vision, natural language processing, and translation. It has been used to re-write an ever increasing amount of the Google codebase, and has won three of the pioneering deep learning researchers the Turing Award, the highest honor in computer science.
Machine learning in biology
The history of applying machine learning to biology has a much earlier starting point than the deep learning revolution. Many researchers used support-vector machines (SVMs) and other types of “classical” machine learning methods to decipher patterns in DNA microarray data long before GPU-accelerated deep learning came onto the scene. With that being said, work at the intersection of deep learning and biology has generated a frenzied flood of activity and interest.
Keeping up with new results in this space can feel like attempting to drink from a firehose. Dozens of preprints and journal articles are released every day describing new models, architectures, and results of using AI in biology. One reason for this is that software is eating AI. That is: new software libraries and free cloud computing resources are now enabling graduate students to train powerful models with very little code (sometimes <100 lines) and prior expertise.
With this surge of activity, it can be challenging to identify which results and advances really stand out and drive the field forward. Žiga Avsec, one of the co-first authors of this new preprint, led a project that moved the entire field forward in 2019. In a collaboration between the Kundaje Lab and the Zeitlinger Lab, the team developed a deep learning model that generated exquisitely high-resolution predictions of how regulatory proteins bind to DNA sequences.
Interestingly, despite the incredible predictive power of the new model and the regulatory biology that was discovered, the group had a challenging time publishing their paper. In fact, Anshul Kundaje ended up publicly voicing his frustration with their difficulty getting the paper published:


Similarly, another talented computational biologist Vikram Agarwal (also co-first author of today’s paper) voiced his own discontentment with academia and left to join Calico Labs, an anti-aging company in California backed by Google.
Solving protein folding
While academic scientists couldn’t seem to reach consensus on the merit of some of the new advances of deep learning in genomics, industry labs charged ahead with research programs aiming their computational resources at unsolved problems in the life sciences.
One of the most notable examples of this has been the entry of the London-based Google DeepMind3 into the field of protein structure prediction. The protein structure prediction problem is the challenge of modeling the relationship between a sequence of amino acids and the corresponding three-dimensional structure of a protein:

This has been widely considered to be one of the foundational problems of biochemistry and computational biology. In order to measure progress on the problem, the Critical Assessment of protein Structure Prediction was founded in 1994 to provide a benchmark for scientists to collectively work towards.
In 2018, the field of structure prediction was dramatically shaken up when DeepMind made their first entry in CASP with AlphaFold. In a performance that felt like the AlexNet moment in biology, AlphaFold took first place at CASP13 by a large margin. This sent a shockwave through the field, which was best described in a blog post entitled AlphaFold @ CASP13: “What just happened?” by Mohammed AlQuraishi.
By 2020 at CASP14, DeepMind’s performance was so impressive even relative to CASP13 that the organizers of the competition issued a press release declaring that the structure prediction problem has effectively been solved.4
A new focus on genomics
Several threads of the story so far intersect when considering the impact of this new preprint from Google. Žiga Avsec is now a Research Scientist at DeepMind. He and fellow DeepMind researcher Daniel Visentin teamed up with Vikram Agarwal and David Kelley from Calico Labs to lead a project that constitutes DeepMind’s first notable entrance into the field of genomics.
The primary motivation for this paper and the broader field of regulatory genomics is to understand the impact of noncoding variation on gene expression. One of the most confounding results from the era of genome-wide association studies (GWAS) has been the incredible amount of association between noncoding sequences of the genome and complex traits. In order to understand how the genome encodes complex cell-type specific regulatory programs, it is crucial to build better models of how DNA sequence composition affects which genes are expressed.
It has been broadly demonstrated that CNNs can be re-purposed to detect regulatory information in DNA when trained on functional genomic assays. In quality work building up to this, David Kelley published an exciting paper that demonstrated the use a deep learning architectural technique called dilated convolutions to model the impact of regulatory sequences up to a distance of roughly 20 kilobases (kb) away from genes.
The major advance in this new work is that the group swapped out the dilated convolutions with self-attention layers, creating what is referred to in deep learning as a Transformer model5, which have been massively influential in the natural language processing community recently. Described at a high level by David, “In these layers, all pairs of elements in your sequence are considered together and decide how much they ought to influence each other and what they have to share. It’s a natural fit for these regulatory DNA tasks, and we’ve been discussing these models for years.” While this seemed like a natural conceptual fit, this took a considerable amount of effort, engineering and computation to successfully accomplish.
Promising progress
The Enformer model (portmanteau of enhancer and transformer) performed very impressively across a wide variety of application domains. One key advance over previous work is that this model extended the range of predictive ability to a 100 kb sequence window around genes, since the input to the model is a 200 kb segment of genomic sequence. The new model outperformed previous architectures on predicting gene expression:

It also seems that DeepMind is not going to rest on its laurels after previous performances at CASP. The group evaluated how well the Enformer model performed on the most recent Critical Assessment of Genome Interpretation (CAGI) challenge:

They accomplished this performance using only the training set that was provided by the challenge. It is interesting to see that DeepMind has focused on this type of benchmark. Given the remarkable progress that was made between CASP13 and CASP14, I am curious to see what the ultimate result will be of DeepMind and Calico’s collaboration to predict gene expression.
Future implications
There are a lot more interesting results in the paper that I haven’t covered, but I want to close with a few thoughts about the implications of this exciting project.
As we have seen from CASP and many other AI challenges, an interesting asymmetry arises between industry and academic labs when scientific problems are reduced to deep learning competitions. Large companies have far more computational resources and highly skilled teams of engineers. This combination enables rapid progress and iteration on problems. In certain fields, it can reach the point where it would be impossible for a graduate student to train a model at the scale that would be necessary to achieve state-of-the-art results.6
This asymmetry is complicated even further by the fact that there is a high demand for deep learning experts in industry, which has resulted in what has been called the AI Brain Drain, where top talent has ended up in industrial labs. It can be even more challenging to stay in academics in biology, when it is challenging to publish cutting-edge models in top journals even after many months of review.
However, despite the complex dynamics, I think that this line of research presents a tremendous opportunity for both industry and academia. In this paper, the authors mention a certain North Star for further progress. This new model led to “a substantial performance increase in tissue and cell-type-specific gene expression prediction correlation from 0.81 to 0.85, one third of the way towards the experimental-level accuracy of 0.94 estimated from replicates”
If we extrapolate from past results at CASP, there is a good chance that we are on the horizon of a world where we have deep learning models that can predict gene expression with the accuracy of experimental replicates.
What will the world be like when that is achieved? It doesn’t matter who gets there first. We could have exquisitely accurate computational models of both protein folding and genome regulation in the next decade. I think that this has enormous implications. This could dramatically accelerate efforts in both protein and genome engineering.
I personally think that this could give rise to the birth of a new computational discipline, concerned with building new tools to perform and utilize new virtual experiments that could rival or exceed experimental accuracy. There is tremendous room in these new pastures for both academic and industrial researchers.
There are already glimmers of this activity as new tools are being developed to aid in the exploration of the predictions of these models. I have recently described some of this here:

However, these exploratory tools only visualize static predictions of these models. There is a tremendous amount of opportunity for new tools to run interactive experiments and perturbations against these types of models.
With that being said, I’ll conclude this week’s highlight of “Effective gene expression prediction from sequence by integrating long-range interactions”. This ended up being a longer post than I will typically aim for, but I think that there was a lot to say and cover about this new model.
I hope that this overview has helped provide a high-level picture of the various aspects of this exciting and fast moving research space. I think that we are only scratching the surface of what we will see in the application of deep learning to genomics in this decade.
If you enjoyed this post, you should consider subscribing! Each week I will highlight a new exciting result in the life sciences.
In the meantime, have a great Sunday!
Here I intend to provide a mile-high overview of note-worthy moments along deep learning’s path from obscurity to the center of computer science. If you are a biologist with some Python programming experience, the free fast.ai courses by Jeremy Howard and Rachel Thomas provide an amazing introduction to machine learning. If you prefer reading, one of my favorite machine learning books is HOML by Aurélien Geron.
DeepMind is probably best known for Alpha Go, the deep learning model that beat the world’s best Go player. This is widely considered to be a seminal advance in AI.
AlQuraishi wrote a subsequent post detailing the implications of CASP14 as well.
For the technically curious in the audience, this blog post provides a nice overview of Transformers, and collects various helpful pieces of information into a readable and visual post.
This section of AlQuraishi’s post eloquently describes this scenario.