
Data-driven virus detection
Automating the development of accurate viral diagnostics

Overview
We have all learned far more about viruses this year than many of us ever expected to learn. COVID-19 has resulted in a large amount of human suffering, death, and economic loss around the world. Our governmental and technical response to this viral pandemic has been extremely unbalanced. Incredibly, synthetic mRNA vaccines are now a topic of household conversation. Unfortunately, the track record for viral diagnostics has been far less impressive.
In February 2020, technical errors in the development and production of a COVID-19 diagnostic caused the United States to fall a month behind on effective testing. This type of delay in testing has enormous ramifications for the effective surveillance of a rapidly spreading viral outbreak. The problem stemmed from the failed design of the PCR test for detecting the presence of the virus. At a certain level, our inadequate technological infrastructure failed us. The head of virology at the University of Washington, Keith Jerome, was quoted saying “primer design is still somewhat of an art, and not fully predictable.”
Last week, we looked at the impressive way in which DNA sequencing, DNA synthesis, and machine learning could be combined to more effectively optimize AAV capsids for gene therapy. The combination of these tools enables the design of viral vehicles that can better evade the immune system during the delivery of therapeutic payloads. What if we harnessed the power of genomics and machine learning to transform diagnostic design from an art into a data-driven engineering discipline?
At the Broad Institute, Hayden Metsky is working on tackling this problem. Hayden is a postdoctoral fellow in the Sabeti Lab, a group that has worked at the forefront of technology development for combatting infectious diseases for many years. I first became aware of Hayden’s work in this space when a probe design tool called CATCH was published in 20191. CATCH applied rigorous algorithm design to the problem of designing probe sets to detect viral genomes from metagenomic data.
Now, Hayden Metsky is the lead author of a new preprint entitled “Designing viral diagnostics with model-based optimization.”2 The study was supervised by Cameron Myhrvold, Michael Mitzenmacher, and Pardis Sabeti. This study combines algorithmic rigor with new genomic data to establish a much more robust approach to developing viral diagnostics.
Key Advances
The main focus of this study is on optimizing CRISPR diagnostics for detecting viruses.3 In order to understand this study, it is worthwhile to think a little bit about how this technology works:
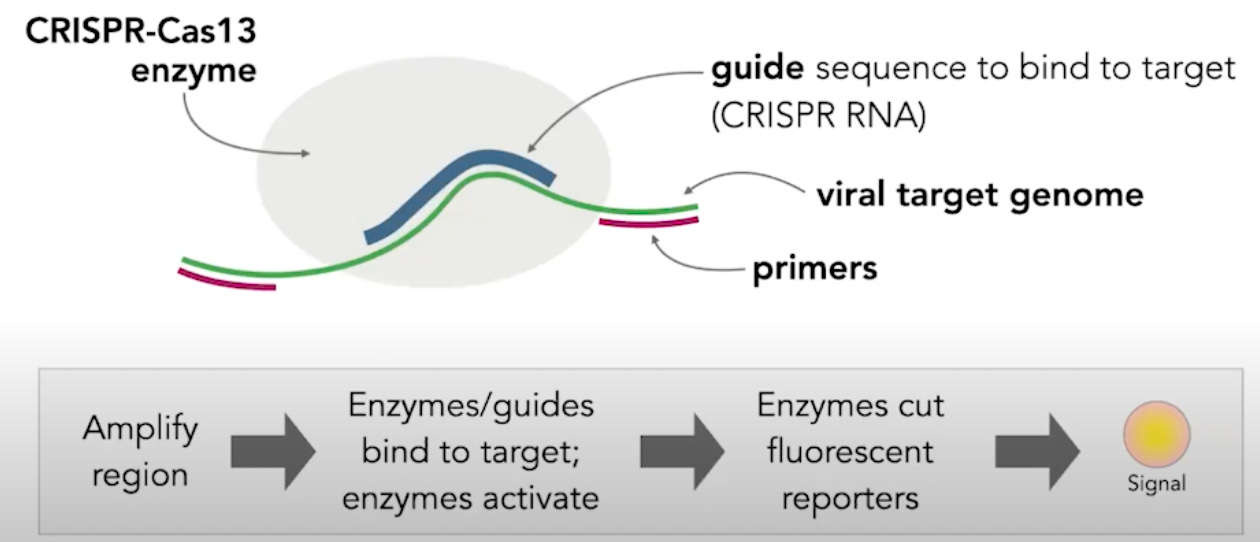
This schematic shows the general workflow of this type of diagnostic. First, a region of a viral genome is amplified, using primers. Next, a guide sequence binds to its target sequence on the viral genome. The guide recruits the CRISPR-Cas13 enzyme, which activates enzymes, ultimately resulting in a detectable fluorescent signal. The fluorescent signal indicates that the target sequence of the guide was present in the sample being tested.
There are considerable challenges associated with choosing the right guides for a CRISPR assay that will make it an effective viral diagnostic.
The authors focus on making progress on three distinct parts of this problem:
1) Improving “predicting a diagnostic’s efficiency in detecting a nucleic acid target.” In the article in the MIT Technology Review quoting Keith Jerome, Neel Patel writes “Even when you have a good database of viral sequences, not all primer sets that look good on a computer will perform well in real life.” In a world where we are accustomed to highly accurate machine learning models that can classify arbitrary images with better performance than humans, this seems wrong. This study set out to change this.
2) Effectively handling viral variation. This is crucial, because “influenza A virus (FLUAV) RT-qPCR tests often have false-negative rates over 10% (nearly 100% on some circulating strains) owing to extensive sequence variation.” The team took advantage of the ability to incorporate this design constraint into their algorithm.
3) Scalability. Ideally, we don’t want to be caught off guard by another viral pandemic and be worrying about failed diagnostics months into the spread of the outbreak. What if we could continuously integrate all publicly available viral genomic data into an automated algorithm that designed highly accurate viral diagnostics on a consistent basis? That is what ADAPT, the software system described in this paper, was built to do.
Results
In order to improve the ability to predict diagnostic performance, this study takes an unconventional approach. Instead of using “thermodynamic criteria and simple heuristics”, the team generated a large library of 19,209 guide-target pairs and measured their fluorescence over time in a multiplexed experiment. They then used this empirical data to build a predictive machine learning model.
This is noteworthy because it generally mirrors progress in other fields. When deep learning first produced dramatic results in the AI field of computer vision, it similarly displaced existing approaches that relied on hand constructed rules and heuristics.
The model performed quite well:
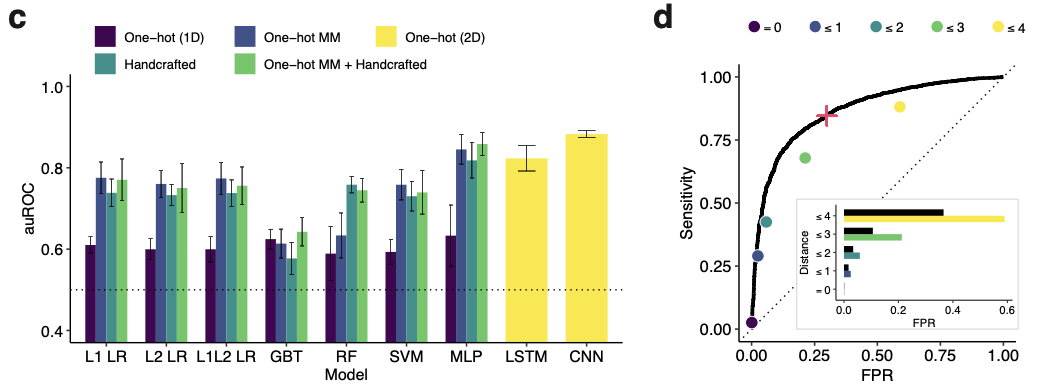
After developing an accurate predictive model, the group set out to develop an algorithm to optimally design a diagnostic assay that takes the sequence variation of viruses into account. They developed a formal objective function for “maximizing the expected activity” of a given probe set on a target region of a viral genome. The proposed algorithm uses locality-sensitive hashing to construct the set of possible probes, and combinatorial optimization to maximize the objective function.
Both the increased predictive performance for diagnostic accuracy and the more rigorous approach to probe set optimization are impressive results for a single study. What I really enjoyed about this work was that the group didn’t stop at this stage. They released ADAPT, a tested, documented, and maintained Python package that automates the entire design process:

This tool is intended to provide an “end-to-end” approach to diagnostic design. The software “automatically downloads and curates data from public databases to provide designs rapidly at scale.” It is also containerized to be able to run in the cloud.
Final Thoughts
Think about how cool this is for a second. This is an automated pipeline for designing highly accurate CRISPR-based viral diagnostics using state-of-the-art machine learning that runs in the cloud. It could have already designed an effective diagnostic the next time we are in a position to need one. If that isn’t a 180 from the testing failure we all suffered through this year, I’m not sure what is.
COVID-19 has impacted the entire world, and one new diagnostic strategy alone isn’t a panacea for all of the ways in which our systems and institutions were unprepared to respond. With that being said, this type of innovative work at the intersection of the computing revolution and the genomics revolution truly has the potential to change the world in meaningful and exciting ways.
Thank you for reading this highlight of “Designing viral diagnostics with model-based optimization”.
If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, have a great Sunday! 🧬
I was also working on computational probe design at the time, but for imaging applications. It was really interesting to see the similarities and differences that emerged between the approaches based on the different goals.
The first version of this study was originally posted on November 28, 2020. Even earlier than that, a prototype of the tool described in this study was used in January 2020 to create assay designs for SARS-CoV-2.
While the focus here is on CRISPR diagnostics, the framework introduced here is computational, and can be applied to other molecular technologies like PCR, as the authors mention.