
Completing the human genome
Using long-read sequencing to resolve the unmapped regions of the genome

Overview
Maps are an essential tool for exploration. They provide a common set of coordinates, enabling navigation and direction. Another key feature of maps is their ability to keep track of where we have already charted, and where we have yet to explore.
In genomics, the foundational map is the reference genome. For each organism that is studied, work begins and ends with the reference genome. For each model organism community, a crucial task is to build this shared resource. A reference genome is created by sequencing and assembling all of the DNA that would be present in a single organism of the species in question.
With this shared set of coordinates, it is possible to establish resources, tools, and annotations that are shared throughout the community. This is a huge part of the story of genome science, and is exemplified by members of the Alliance of Genome Resources such as WormBase and FlyBase.
This general strategy is also a core aspect of human genomics. In order to construct the consensus map of the human genome, an international project called the Human Genome Project ran from 1990 to 2003 in what amounted to a herculean effort to map out all of the bases that constitute a single human’s DNA.
Another team from a corporation called Celera Genomics pursued the same goal in parallel to the HGP, ultimately leading to a race to the finish. The result was a joint release of the first draft of the human genome by Celera and the HGP in 2001. It is worth noting that the cost of completing the project was nearly 3 billion dollars. Since the completion of this project, genome sequencing has followed a cost curve that has outpaced even Moore’s Law in computing:

The development of a reference genome was absolutely critical for progress in human genomics, and was of central importance in the sequencing revolution, serving as a foundational tool for sequencing alignment methods as well as genome assembly methods.
Despite this enormous milestone, our maps have actually still contained uncharted territory. The initial draft of the human genome and all following patch updates have consisted of the euchromatic regions, which comprises roughly 92% of the genome. These regions of the genome are less compressed and are enriched for genes and transcriptional activity. The remaining 8% of the genome absent from our current reference is made up of more tightly condensed heterochromatic regions which make up a large portion of centromeres and telomeres, and other complex and often repetitive regions1 which are very difficult to accurately map.

In order to move beyond this, the Telomere to Telomere (T2T) Consortium was established by Adam Phillippy, Karen Miga, and Evan Eichler with the ambitious goal of generating the first complete assembly of a human genome. The open and highly collaborative T2T consortium has been enormously productive, having already published complete assemblies of the human X chromosome and chromosome 8.
Now, the T2T consortium has posted a preprint: “The complete sequence of a human genome” which marks a major milestone for the field of genomics. These results have generated an enormous amount of excitement in the genomics community, and have considerable implications for the field moving forward. I’ll highlight this amazing work today, and look at what this might mean for the future of genomics.
Key Advances
To really appreciate the results of this new work, I think it is worth highlighting the contrast in methodology between the original HGP and the T2T consortium. This preprint details the previous strategies in the following way:
Unlike the competing Celera assembly, and most modern genome projects that are also based on shotgun sequence assembly, the GRC human reference assembly is primarily based on Sanger sequencing data derived from bacterial artificial chromosome (BAC) clones that were ordered and oriented along the genome via radiation hybrid, genetic linkage, and fingerprint maps.
Frankly, genomics and molecular biology have moved so rapidly as disciplines that these techniques haven’t been a large part of my scientific education or career2, but I’ll dig into this workflow a little bit here.
The idea here is take a portion of the genome3 and insert it as a vector into a bacteria (typically E. coli, the workhorse of molecular and synthetic biology), reducing the challenge of sequencing into a smaller subproblem. With the BACs established, Sanger sequencing is a protocol that predates shotgun sequencing and is a gold-standard benchmark for the accuracy of newer methods. It proceeds by continually introducing fluorescently labeled di-deoxynucleotide triphosphates, to detect each base of the template sequence at a time.
With the BACs sequenced, the listed techniques (radiation hybrid, genetic linkage, fingerprint maps) are all labeling or analysis techniques to stitch together the separately sequenced contigs into scaffolds of chromosomes, ultimately reconstructing the complete genome.

While this approach built the foundation for human genomics, there was room for improvement:
This laborious approach resulted in what remains one of the most continuous and accurate reference genomes today. However, reliance on these technologies limited the assembly to only the euchromatic regions of the genome that could be reliably cloned into BACs, mapped, and assembled.
There is an analogy that can be drawn here to machine learning, where the engineering and data generation necessary to transition from a minimally viable model to a model robust enough to handle tricky but important edge cases can be enormous. In some cases, it can require the development of completely new tools and strategies.
This is also the case here. To dramatically reduce the labor of assembly and mitigate the systematic inability to profile heterochromatin, the T2T consortium turned to newer sequencing technologies. In particular, they “leveraged the complementary aspects of PacBio HiFi and Oxford Nanopore ultra-long read sequencing” which both generate reads that are much longer than shotgun sequencing.
On a conceptual level, this dramatically reduces the complexity of the assembly problem, because individual reads can be long enough to entirely span regions of the genome that were previously impossible to correctly map, and these large reads make it much easier to stitch together contigs and scaffolds. Less puzzle pieces to place!
Another key strategy choice of the T2T consortium was to choose the CHM134 cell line, which is derived from a complete hydatidiform mole (CHM). This is a type of molar pregnancy where a sperm combines with an egg that has lost its DNA, resulting in a growth that is not a viable pregnancy. One artifact of this process is that the resulting cells are typically 46,XX (as CHM13 is), with both copies of all chromosomes coming from the same sperm. This means that they are “almost completely homozygous and therefore easier to assemble than heterozygous diploid genomes.”
Between long-read sequencing and this effectively single-haplotype cell line, the T2T consortium had the tools and strategy in place for “the construction, validation, and initial analysis of the first truly complete human reference genome” which is an incredible accomplishment.
Results
Without further ado:
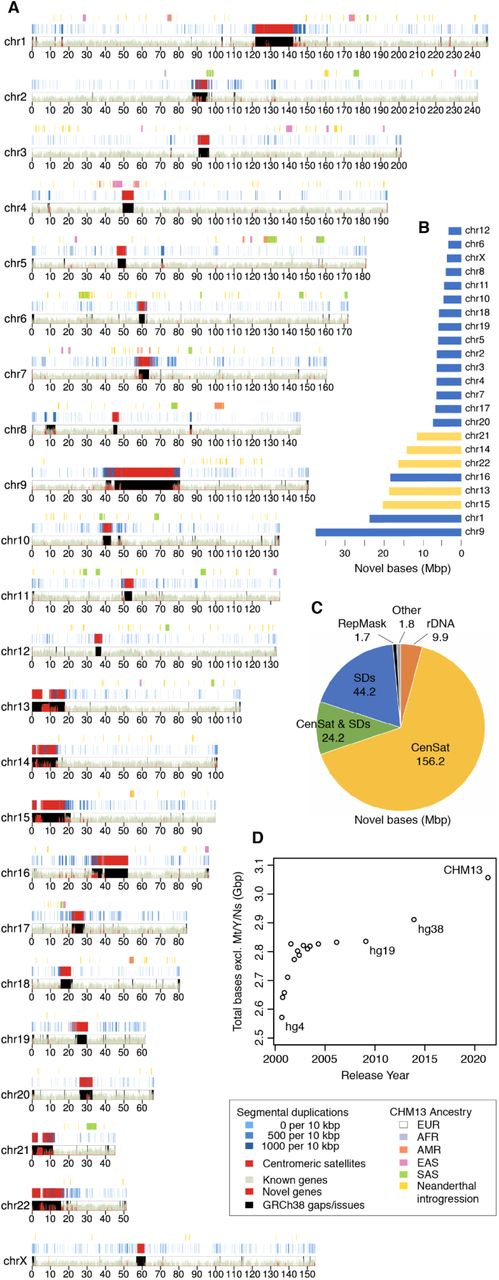
This is a karyoplot5, which represents all of the coordinates of the genome. A key point to pick up from this visualization is the marked areas that are novel genes or the centromeric satellites (red), and the resolved gaps from the previous assembly (black). Viewed in this way, it is clear how much of our genomic map was left uncharted before this work!
As I’ve mentioned, the long reads are a game changer for assembly. Here is how this process works:
The basis of the T2T-CHM13 assembly is a high-resolution assembly string graph built directly from HiFi reads. In a bidirected string graph, nodes represent unambiguously assembled sequences and edges correspond to the overlaps between them, due to either repeats or true adjacencies in the underlying genome.
This is really elegant and exciting from a bioinformatics perspective. Graphs are simultaneously simple but flexible and powerful representations of data that are extensively studied and utilized in computer science. There is a mature set of tools for working with graphs, and thinking about them.6
After an initial automated construction, the graphs underwent rounds of manual curation and polishing to resolve certain tangles that could be be simplified with careful inspection to be represented as unique walks through the string graph. In areas where it was challenging to resolve ambiguities in the assembly, Oxford Nanopore reads were aligned to provide additional evidence.
Here is what these graphs look like:

Pretty beautiful! In some of the zoomed in regions like the centromeres and rDNAs, these graphs provide some visual intuition for how challenging these parts of the genome would be to assemble when working with smaller puzzle pieces. For these highly complex and repetitive regions of the genome, it is nearly impossible to map them correctly without sequencing technology that can generate reads long enough to span them entirely.
An absolutely essential part of establishing a new assembly is to validate that it accurately models the underlying sequence data and the biology that those data are measuring. All of the work from the T2T consortium has been really rigorous and high quality with respect to this issue, and it is taken very seriously. I’m not going to dive into this data here, but genome biologists considering using the T2T-CHM13 assembly should absolutely read this section of this preprint. As somebody who uses rather than assembles reference genomes, I have learned a lot about validation strategies from this work, and I have found the description to be very accessible.
I want to highlight one more key aspect of this work and how it has been shared that is essential to building a trustworthy community resource: all results are hosted on AWS and linked to from this Github repository. This type of open science approach was a really critical aspect of how T2T structured collaboration across the consortium, and fostered an incredible feedback loop that made the final result more robust. At the end the end of the day, it is incredibly cool that there is an open repository with the description: “The complete sequence of a human genome.”
Final Thoughts
A crucial aspect to realize about making improvements to the reference genome is that it has tremendous downstream impact for research and engineering in genomics. Because it is such a foundational coordinate system, it impacts everything that relies on it. This means that all new sequencing data can be more accurately mapped with a complete reference. All searches for off-target CRISPR sites could be more comprehensive and more accurately account for all of the genome. In the past I have worked on computational probe design for imaging applications, which could be made more accurate and effective with a more complete reference genome. Nearly all areas of bioinformatics stand to benefit from a more comprehensive map of the genome.
Even now looking at the first complete human genome assembly, the authors spoke of a future for genomics that moves beyond the concept of a single reference genome. Given the rate of technological progress in sequencing and assembly, “a collection of high-quality, complete reference haplotypes will transition the field away from a single linear reference and towards a reference pangenome that captures the full diversity of human genetic variation.” This is a really inspiring vision for a near future where we could have “the routine and complete de novo assembly of diploid human genomes” which is incredible to think about. This would establish an entirely new frontier for scientific discovery, just as the original reference genome did.
A really inspiring aspect of the T2T consortium has been the sense of exploration in the face of challenging genomic complexity. Despite the historical inability to accurately resolve these regions, the T2T researchers have remained undaunted, pushing forward with new technology and a collaborative spirit.
To reflect on this, I’ll close with a quote by a scientist who I’m sure would be absolutely delighted by this work:
It is like a voyage of discovery into unknown lands, seeking not for new territory but for new knowledge. It should appeal to those with a good sense of adventure.
- Frederick Sanger
Thanks for reading this highlight of “The complete sequence of a human genome”. If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, have a great Sunday! 🧬
Some of these really tough to map regions include segmental duplications, ribosomal DNA (rDNA) arrays, and satellite arrays.
As a young scientist/engineer, I’ve been trained with the pampered luxury of QIAGEN kits and Jupyter notebooks!
Roughly 150,000 basepairs in length, or 150kb.
Which I admittedly always first read as “chromosome 13” in my head.
This figure was created with an R visualization package called karyoploteR. As the author of an R package for genomics visualization, it does bring me a general degree of satisfaction that this is Figure 1. R is great!