


Visualizing cellular trajectories
A new computational tool for analyzing changes in single-cell gene expression

Visualization is more than a tool for finding patterns in data. Visualization leverages the human visual system to augment human intellect.
Overview
Few trends in the life sciences have been more pronounced than the adoption of single cell sequencing technologies:

As the cost of next-generation sequencing dropped exponentially after the completion of the Human Genome Project, it became possible to move from sequencing cells in bulk to collecting measurements from individual cells in a sample. This change in resolution is essential for capturing dynamic processes in complex tissue microenvironments. The classic example for describing the advantage is to consider a smoothie vs. fruit:

Although single-cell transcriptome sequencing (scRNA-seq) is now the gold standard for measuring gene expression profiles, it still comes with limitations. One of the technology’s largest shortcomings is that the data represents a static snapshot of a dynamic process. The RNA transcripts that are measured represent a single point in time, like a single frame from a movie.
One of the most impactful new analytic tools for better capturing cellular dynamics from scRNA-seq data has been RNA velocity, which accurately predicts the change in gene expression from the static snapshot measured on the timescale of hours. This type of prediction is a huge enabler for studying complex processes that unfold over time and space, such as organismal development and disease progression.
There is one critical methodological question that arises with this technique: how do we build any intuition for the biology that is represented by a high dimensional vector of the time derivative of gene expression? The wonderfully creative inventor Buckminster Fuller compared aspects of modern science to flying a plane blind: “Their invisible procedures thenceforth to this day have a counterpart in modern air transport and night fighter flying—which is conductable and is usually conducted entirely ‘on instruments’.”
Flying bind does not mirror the way in which we learn about the physical world around us. Humans are fundamentally visual creatures, with a greater proportion of our brains and cognitive processes devoted to visual perception relative to our other senses. We are hardwired to detect visual patterns, and as the data visualization pioneer Mike Bostock points out, “visualization leverages the human visual system to augment human intellect.”
Today, I’m going to highlight the new version of a preprint which makes progress on this important problem: “VeloViz: RNA-velocity informed embeddings for visualizing cellular trajectories”, led by Lyla Atta from the JEFworks Lab at Johns Hopkins University. This is the first project from the lab, which was established by the talented researcher Jean Fan who contributed to several cutting-edge spatial transcriptomics projects including the initial RNA velocity project. I’m sure that this is only the beginning of the exciting work coming from this group.
Key Advances
When thinking about this tool, the general workflow for studying the trajectories of single cells can be thought of in three phases. First, the expression profiles of the cells in a sample have to be measured with scRNA-seq, and the data has to be processed. Once the static snapshots of cell states have been collected, they are computationally expanded into vectors1 using the RNA velocity technique. Finally, these vectors need to be converted into a human interpretable format, which involves embedding them in a lower dimensional representation where the patterns are visually interpretable.
VeloViz is a new computational tool that implements a new and more accurate approach to generating “RNA-velocity-informed 2D and 3D embeddings from single cell transcriptomics data.” The algorithm has a series of steps for going from RNA velocity predictions to interpretable embeddings:

The formulation of composite distance in step two incorporates two key pieces of information for each pair of cells in the data: 1) transcriptional dissimilarity, and 2) velocity similarity. By computing the distance using these two aspects, it will be “be minimized when Cell A’s predicted future transcriptional state is similar to Cell B’s observed current transcriptional state and when the direction of Cell A’s RNA velocity is similar to the direction of the transition from Cell A to Cell B.”
With this approach to computing distance that takes into account the underlying biology, the remaining steps involve using the distance metric to create a graph, pruning the graph to remove any incorrect nodes, and then visualizing it in 2D or 3D2.
Results
Enough talking about these graphs, let’s see what an example looks like:

This is pretty beautiful. This is scRNA-seq data from Bergen et al., 2020, and represents the formation of the endocrine component of the pancreas during development. You can see a cycling population of ductal cells, and the transition between various progenitor cells during development before final differentiation.
What I really enjoy about this type of visualization is how intuitive the interpretation is. scRNA-seq is a fundamentally complex measurement technique, relying on the successive use of microfluidic devices and next-generation sequencing. RNA velocity introduces a computational layer to derive more useful information from the initial measurements. After all of this molecular and computational work, I love to see the data embedded in a representation that is so tangible. The cells are flowing between states, separating and differentiating. These are patterns that our eyes have evolved to register, and represent meaningful biological events.
This is not the first technique that has been used to embed RNA velocity data. This paper provides a detailed comparison to a set of other common techniques: PCA, t-SNE, UMAP, and diffusion maps:

Overall, the results show that VeloViz consistently outperforms existing techniques on the metric used for determining the accuracy of the resulting embedding3. One interesting set of results in this work is that VeloViz showed better performance on datasets with missing intermediate cell states.
In thinking about this, the authors said “We hypothesized that incorporating information about each cell’s predicted future transcriptional state could enable VeloViz to more robustly construct representative cellular trajectories even when the sampled cell states contain missing intermediate cell states or gaps in the underlying trajectory.” This difference can be understood visually:
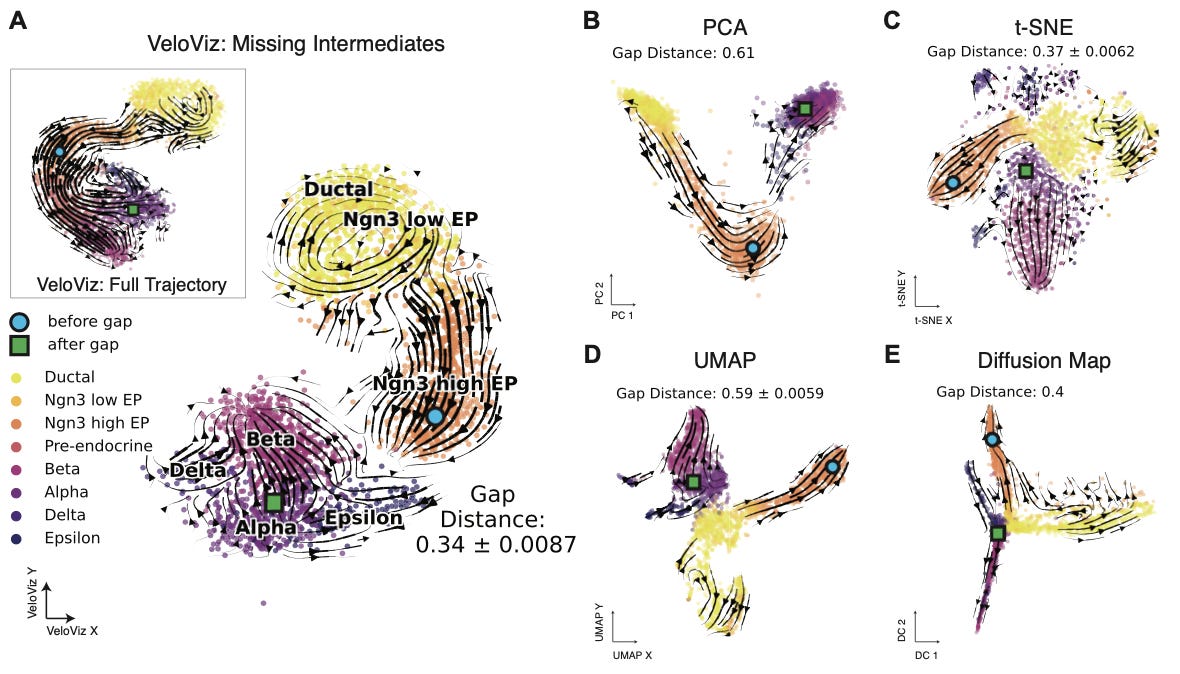
When gaps were introduced into the data, VeloViz was the only embedding technique that was able to durably capture the full trajectory in the dataset. In particular, you can see how the pocket of cycling ductal cells only shows up in the VeloViz embedding.
Final Thoughts
This concludes my highlight of “VeloViz: RNA-velocity informed embeddings for visualizing cellular trajectories” from the JEFworks Lab. If you are a single-cell biologist, you should consider checking out their tutorials and adding this new tool to your visualization toolbox. If you aren’t a single-cell biologist, I hope that this was an interesting primer on this field, introducing you to some of the analytical techniques that are used and the ways that the data are visualized and interpreted.
If you are a scientist (or aspiring scientist) and this work really speaks to you, the group has listed a ton of opportunities for joining their team, ranging from the high school level to the postdoc level.
If sequencing is to be considered “the broadly enabling microscope” of the 21st century, perhaps this type of computational tool represents the light that illuminates the sample, flooding our visual field with patterns from a hidden world.
If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, have a great Sunday! 🧬
Again, these vectors represent predicted change in gene expression over time.
This step is flexible. With the graph in hand, it is possible to use a variety of approaches to create the final graph layout. Examples include force-directed graph drawing or UMAP visualization.