
Human Pangenomics
The v1 release from Human Pangenome Reference Consortium

Welcome to The Century of Biology! Each week, I post a highlight of a cutting-edge bioRxiv preprint, an essay about biotech, or an analysis of an exciting new startup. You can subscribe for free to have the next post delivered to your inbox:
Enjoy! 🧬
Today’s Century of Biology post is brought to you by… Cromatic

The biotech industry is evolving. Launching a new startup has become less capital intensive. A new generation of founders are building nimble teams, and outsourcing portions of their R&D to stay lean and focus on core scientific competencies.
Outsourcing can come with headaches. It can be challenging to decide on the right vendor, compare prices, and track project progress. While contract research organizations (CROs) are an important part of the modern biotech ecosystem, these partnerships come at a cost.
Cromatic is on a mission to help modern biotechs make the most out of outsourcing. Their vision is to develop the infrastructure necessary to make contract management a painless, daresay enjoyable, experience. Cromatic makes outsourcing easier than ever so you can focus on the science.
Cromatic is currently performing outsourcing searches for early stage biotech companies for free. Learn more here.
Sometimes I feel science FOMO. The number of exciting frontiers always exceeds what one individual can explore in a given lifetime. I’ve always admired advances in physics and aerospace engineering—rockets are extremely cool. There are fascinating problems in oceanography, climate science, and ecology. Computer science is also full of beautiful research problems, both theoretical and applied.
After a bit of daydreaming, I remember how incredibly lucky I am to be a geneticist in the 21st century. As a scientist, I get to experience the benefit of the absolutely ridiculous sequencing cost curve that I’m continually citing. Our ability to measure life’s hereditary material has expanded dramatically in the new millennium—opening the door to totally new areas of exploration and technology development.
After a brief lag in cost decline around $1000 per sequenced genome—a feat that initially cost $3 billion—the field of genomics has entered into another phase of accelerated innovation. In my view, this boils down to two primary components: 1) a surge in market competition for sequencing technology, followed by 2) a wave of academic developments leveraging new tools to make meaningful advances.
Today, I’m going talk about a new set of exciting results in the second bucket. The Human Pangenome Reference Consortium (HPRC) has recently released the first draft of their new reference. I’m going to dive into the implications of this work for genomics.
Before I do, I want to cover a bit of housekeeping. In my current formulation of this newsletter, I’m aiming to talk about three things: 1) what is happening in genomics and biotech, 2) how it is happening, 3) and why it’s important that we continue to expand and accelerate the sector.

I view these as three adjacent steps on the staircase of abstraction. My recent essay, Viriditas, is an example of the why category. It is a brief excursion into philosophy and a call to think carefully about the future that we want to build. When I talk about companies like Ginkgo, or analyze specific technology stacks, this is an attempt to explain what is happening in the biotech revolution. Finally, my other essays highlighting specific scientific results like the new plant genetic circuits being developed are an attempt to describe how some of this new technology actually works.
There is going to be a difference in the level of technical detail between each essay. I will spend more time expanding on the specifics in order to effectively convey the core ideas of important new results. I’m aiming to support highly curious readers with enough background for the story to make sense, while keeping the newsletter a valuable information resource for practicing scientists. This isn’t an easy task, but I hope to continue to get better at it.
With that being said, there are some incredible scientific stories to tell. For example, I was really proud to cover the completion of the human genome this past year, and to interview one of the scientists leading the project. The timeline of coverage was also interesting:

The preprint describing the work was posted on bioRxiv on May 27th, 2021. In the subsequent two weeks, I posted my essay and interview. At the end of the following month, the award-winning science journalist Carl Zimmer published a piece about the work in the New York Times. Nearly a year later, the peer-reviewed article was published in Science, with an accompanying piece in TIME Magazine. In May 2022, it was announced that Adam Phillippy, Karen Miga, Evan Eichler, and Michael Schatz were in the TIME list of “100 most influential people of 2022” for their scientific contributions.

In other words, if you were a COB subscriber during this time, you were a genomics hipster! You would have been thinking about this cutting-edge science nearly a year before it went mainstream. Oh, you’re excited about the completion of the human genome? Old news.
Even with all of this fanfare, these scientists never missed a beat. They are members of a new group called the Human Pangenome Reference Consortium (HPRC) that is continuing to reshape genomic technology. Recently, I covered the HPRC’s cutting-edge work on automating genome assembly. Now, it’s time to explore the most recent development: the release of the first draft of the human pangenome reference.
We’re going to take the following path to get there:
A brief background on reference genomes
Where the T2T project took us
The new pangenome draft
Implications for genomics
Let’s jump in! 🧬
A brief background on reference genomes
The completion of the Human Genome Project (HGP) marked the beginning of the field of human genomics as we know it. What did it actually mean to complete the HGP? What was the scientific result?
The HGP was a massive global effort to produce the first reference map of human genetic information. All of our genomes are overwhelmingly more similar than they are different. Differences are a rounding error—only 0.1% of the bases will differ in a given individual.
Given our underlying genetic similarity to each other, it’s possible to construct a reference genome that represents the shared stretches of DNA that comprise each human chromosome—this totals roughly 3 billion nucleotide bases. In genomics, variants are represented as bases that differ in a given individual relative to the reference sequence. For example, at a position on chromosome 21, there is a G instead of a T.
So how is a reference genome constructed? In the big picture, there are two important steps. First, stretches of DNA have to be empirically sequenced. The HGP spurred huge developments in high-throughput sequencing technology, which I’ve talked about here. Second, the individual sequencing reads need to be assembled into larger and larger stretches to fully cover individual chromosomes.

{kind=link}
This is part of the beauty of genomic technology: it blends molecular biology and computer science to achieve absolutely incredible results. Modern sequencing instruments simultaneously sequence millions of fragments of DNA, and complex assembly algorithms stitch them together.
While the completion of the Human Genome Project was an enormous accomplishment, many problems were left on the table. To start, substantial portions of chromosomes weren’t actually finished because of technological limitations at the time. The initial reference was also singular by definition: it only contained one reference sequence for each chromosome—limiting our ability to accurately represent genetic variation as we will see. Despite these warts, the initial reference genome was a defining resource that made the birth of human genomics possible.
Where the T2T project took us
It’s hard to overstate the centrality of reference genomes within genomics. They underpin the majority of analysis that takes place within individual projects. When sequencing is used as a measurement technology, it is normally not used to do de novo assembly. Instead, the sequence reads are mapped to their position in the reference—like a lookup operation in a dictionary. Cheap sequencing and a quality reference genome have enabled scientists to ask an enormous variety of questions about gene regulation, genetic variation, cancer, immunology, development, you name it.
While the field took off, our reference map remained incomplete. This led to an inherent bias in analysis: you could only study regions present in the existing map. A new generation of genome explorers became increasingly fixated on the complex unmapped regions of chromosomes. This remained a massive technical bottleneck, until there was a new opening.
High-throughput sequencing is often referred to as next-generation sequencing (NGS), because of the enormous advance they represented relative to previous approaches. Now, a new class of sequencing technologies has earned the label of third-generation sequencing. This term is used to refer to long-read sequencing technologies, of which PacBio and Oxford Nanopore are the most prominent. With these new technologies, a group of researchers realized they had the necessary ingredients to finally complete the genome.

{kind=link}
When adding a bit of detail to the picture of how assembly works, the importance of read length becomes clear. Reads are placed into contigs—short for contiguous—in instances where points of overlap between reads are clear. These contigs are placed into scaffolds, which can take manual curation or additional types of evidence like optical mapping. When starting with longer individual reads, the assembly process becomes much less complex—analogous to a puzzle set with a smaller set of pieces. It also becomes possible to span entire regions that are practically impossible to correctly assemble with a single read.
Armed with third-generation sequencing technologies, the leaders of the Telomere-to-Telomere (T2T) project organically formed a decentralized network of scientists collaborating to finally complete the genome. Coordinating across the country using a massive Slack workspace, the group accomplished their goal and created the first gapless human genome assembly. For a deeper dive on their seminal preprint, you can check out my earlier post about it here.
The new pangenome draft
After my interview with Karen Miga, it became immediately clear to me that the T2T project wasn’t an endpoint; it was a milestone in a much broader vision for the future of genomic technology.
The goal was to complete the first gapless genome. The meta-goal was to advance technology to the point that the idea of a single reference genome would be obsolete.
In a recent conversation with a computer scientist, we reflected on the importance of the gaming industry for the field. It has served as a test bed for engineers like John Carmack to innovate on graphics, helped AI scientists to train their RL agents, and led hardware engineers to develop GPUs. Tangible problems and fast feedback loops enable world-class engineering.
In the same way, the T2T project has served as a springboard for a number of more general advances for the entire field. A lot of new tools and infrastructure need to be developed to achieve the first gapless assembly. What if we didn’t stop at one complete assembly? What if we used these new tools to develop multiple complete genomes, and developed new ways to integrate them together to more accurately represent the entire human race?
This is the vision of the Human Pangenome Reference Consortium (HPRC) that I mentioned earlier. The T2T scientists are key members of this group, along with many of the top minds in the field. One of the primary goals of the HPRC is to move beyond the reference genome, by creating a pangenome reference that integrates the genomes of 350 individuals with diverse ancestries—which would be expected to represent most common genetic variation in the human population.
Obviously, this will require a lot of technological progress to accomplish. I recently wrote about the HPRCs work on automating genome assembly, which will be essential in order to generate hundreds of individual reference genomes.
Once you have a large number of complete genomes, how would we integrate them? Here, the idea is to use a new foundational data structure for genomics: the sequence graph. Succinctly described, this is a graph “in which nodes correspond to segments of DNA. Each node has two possible orientations, forward and reverse, and there are four possible edges between any pair of nodes to reflect all combinations of orientations.”
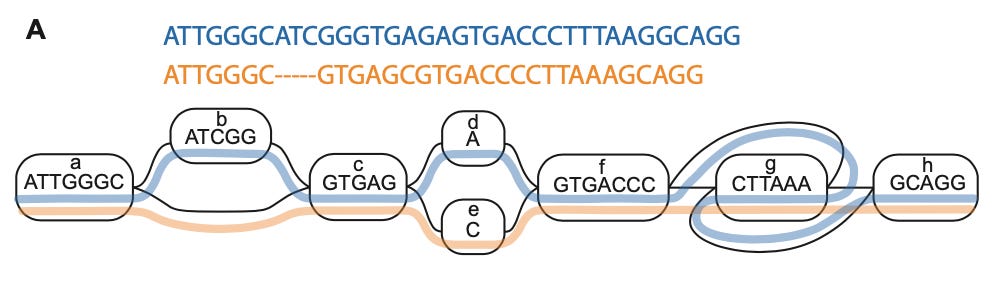
Visually, this resembles a subway map where genetic variation is represented as different routes along the graph. By moving from a single path—which is the idea of a linear reference genome—to a graph structure, we can much more accurately represent the genetic variation present in the human population. This is what is now called the pangenome.
The recent preprint from the HPRC describes their first draft of this new reference. The building blocks for the new graph are “47 fully phased diploid assemblies from genomes selected to represent global genetic diversity” that were assembled in this study. Again, this is a massive testament to the rapid progress in sequencing technology and assembly algorithms.

In this figure, we get a visual impression of the quality of the new assemblies, where “Regions flagged as haploid are reliable (colored green), on average they constitute more than 99% of each assembly.” All of the new genomes were also annotated, which is a ton of work that I’m not going to cover here.
With these new assemblies, they evaluated three different graph construction methods for building the pangenome (Minigraph, Minigraph-Cactus, and PanGenome Graph Builder). Seeing as Minigraph is another tool written in pure C code by Heng Li (who is an author), it looks like my favorite genomics meme will continue to ring true in the pangenome era:

With that being said, let’s zoom out and think about what a pangenome future looks like for the field of genomics.
Implications for genomics
Most people are familiar with the phrase “a rising tide lifts all boats” which nowadays is often used in economics. It’s meant to convey the concept that there are certain properties that are so fundamental that if they are improved upon they will have downstream benefits.
In technology, this can certainly be true. I remember being intrigued by this idea when I watched an interview of Sergey Brin where he talking about how investing in fundamental layers of technology could have “a multiplied effect” on many different applications. CRISPR has accelerated all fields of biology. Fundamental ML breakthroughs lead to better self-driving cars, language models, and diagnostic tools. As both a scientist and investor, I’m obsessed with these types of tools.
I think that the new pangenome reference is this type of advance. There is a section of the preprint entitled “Applications of the pangenome” in which they demonstrate substantial advances for short variant discovery, structural variant genotyping, represent tandem repeats, RNA-seq mapping, and ChIP-seq analysis in a single paper. All of these different types of genomic analyses were made more accurate by making the transition from a single linear reference genome to a pangenome.
So what will this work end up impacting? All of genomics. Whether it is large-scale population studies of human genetic variation, personalized genomics studies of individual genomes, or the types of platform technologies based on sequencing and synthesis that I often write about. Already, “Making the switch to using pangenome mapping is not significantly more expensive computationally, and resulted in an average 34% reduction in errors vs. using the standard reference methods.”
Admittedly, there are going to be some serious growing pains, including ones that aren’t scientific in nature. Adoption doesn’t happen overnight. Educational resources will need to be developed, and better tools, data structures, and file formats will need to be written. There will also be regulatory and logistical hurdles. The most widely used current reference genome is called GRCh38. I’ve been told many times that the previous reference, GRCh37, is still in wide use in medical and clinical genetics because of the associated difficulty of transitioning existing workflows.
As these kinks are ironed out, I’m confident that pangenome variation graphs are going to play an important role in the future of genomics. I, for one, can’t wait to see the discoveries that they unlock.
Thanks for reading this week’s highlight of the new pangenome reference created by the HPRC. If you don’t want to miss the next preprint highlight, biotech essay, or startup analysis, you can subscribe to have it delivered to your inbox for free:
Until next time! 🧬
Having another, better human reference genome is especially important in clinical laboratories, but only if adopted and implemented in practice. I haven't found any policy recommendations for upgrading the reference genome in a clinical lab. Adopting this newest reference could be decades out without a published policy position by respected organizations.