
Ejecting molecules from nanopores
Advances in targeted nanopore sequencing

Overview
Nanopore sequencing is absolutely incredible. While much of the next-generation sequencing (NGS) revolution in genomics has been driven by “short-read” sequencing technologies such as Illumina, “long-read” approaches such as nanopore sequencing have emerged as powerful new platforms with many benefits.
Nanopore sequencing determines the bases of a read by changes in the ionic current as a DNA or RNA molecule is driven through a nanopore:

One considerable advantage of nanopore sequencing is the difference in read length achieved. Nanopore currently holds the record for the longest read, at a whopping 2.3 Megabases (Mb)1! This provides a considerable advantage for mapping and polishing complex regions of the genome that have remained unmapped with conventional short-read sequencing.
Another benefit is the Star Trek level of coolness of the sequencing instruments themselves, which are essentially sophisticated laptop dongles:

Having just finished the Mars Trilogy by Kim Stanley Robinson, I can picture future Martian explorers wielding these types of instruments, and it still blows my mind that this exists in our lifetime. These devices enable sequencing in areas previously unimaginable, such as directly at the site of field experiments.
Despite all of the advantages, one of the largest drawbacks for nanopore sequencing has been a lower sequencing accuracy. However, there have been tremendous improvements in both the chemistry and in the software, with improvements such as high accuracy neural network “basecalling” algorithms.
All of these advances have set the stage for talented computational genome scientists to solve new methodological problems. Johns Hopkins University has been an absolute powerhouse in computational biology, with the Salzberg, Schatz, and Langmead labs among many other talented groups. Collectively they have produced incredible tools such as the Bowtie and Bowtie2 (which has greatly benefited my own research) short-read aligners, and Tophat for splice alignment, just to name a few.
Whenever I see a new paper coming out of one of these groups, I make sure to take a look! Their methodological rigor, careful evaluation, and clear presentation always have me taking notes.
For my first Century of Biology post, I am going to highlight their recent bioRxiv pre-print “Pan-genomic Matching Statistics for Targeted Nanopore Sequencing”, which details an exciting new method for working with nanopore sequencing data.
Key Advances
One amazing new aspect of nanopore sequencing is that data can be read from the device in real time, and there is a software API for ejecting molecules from the nanopores, which can enable more targeted sequencing. This reminds me of George Church’s quote that biology is “the nanotechnology that really works.”
I first learned about this method when the Schatz Lab published their Nature Biotechnology paper describing UNCALLED, a new mapping software leveraging the Oxford Nanopore Technologies (ONT) Read Until API. Now, with contributions from UNCALLED first author Sam Kovaka and Michael Schatz, this new work led by Omar Ahmed and a talented team describes SPUMONI: a new method for nanopore read classification.
One area where UNCALLED required further performance improvements was in the classification of reads when the reference sequence is highly repetitive. SPUMONI aims to fill this gap. According to the authors, “SPUMONI’s core insight is that a read’s matching statistics (MSs) with respect to an index can reveal whether it has a “good” (i.e. long, high identity) approximate match to the index, without having to perform a more costly read alignment.”
In order to compute matching statistics instead of an expensive read alignment, they extended their previous method accepted at RECOMB 2021 called MONI, by making it faster and geared towards use with the Read Until API for rejecting reads. I will leave a more exhaustive description of the method for interested readers to explore their very approachable paper.
Results
As I mentioned, the Langmead Lab and co. at JHU provide very careful evaluations of their tools, which helps to inform scientists interested in adopting them. This paper is no different.
I will share one exciting result from the paper. Microbiome research has benefitted tremendously from the development of nanopore sequencing, where it is often the tool of choice for metagenomics studies. The authors used a recent ONT dataset from a microbiome study, and filtered out the reads that were likely to be human. Next, they combined it with a set of simulated human reads with a mean read accuracy of 90%.
Here is what they saw:
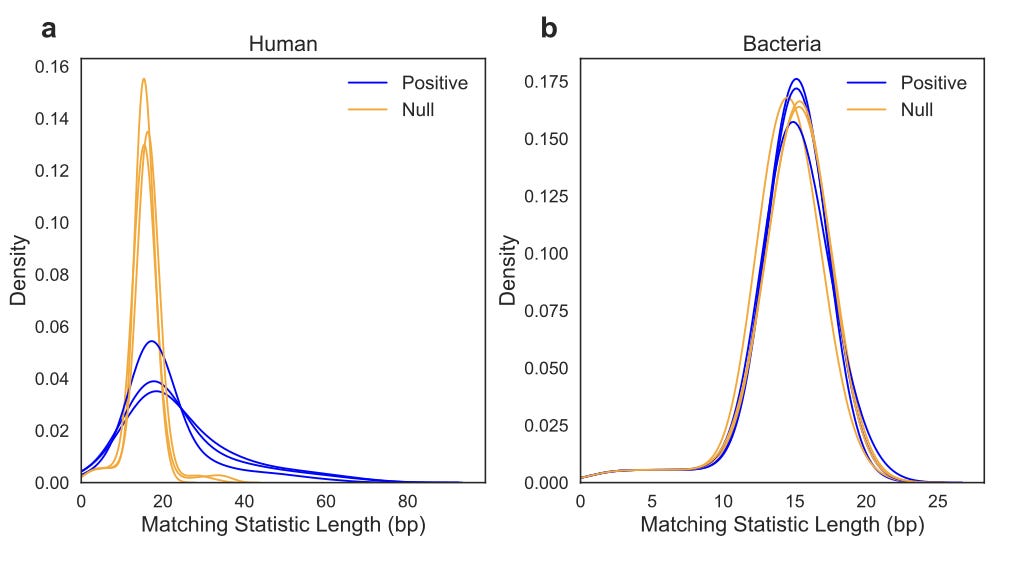
Each curve represents the first ~1.6 Read Until seconds of three randomly selected reads, which ends up being 720 bases. A greater divergence from the null distribution indicates that the reads are matching the positive index, or being filtered.
What does this mean at a high level? This is an example of how for a microbiome study, SPUMONI could be used to classify and eject reads that are likely to be human DNA from the nanopore, leading to targeted enrichment of bacterial reads. In real time. Pretty incredible!
Final Thoughts
I hope that you’ve enjoyed this highlight of the pre-print “Pan-genomic Matching Statistics for Targeted Nanopore Sequencing” from the Langmead Lab at Johns Hopkins. If you’re an aspiring researcher that found this science really exciting, consider reaching out to their group. They have a listing of open positions here.
If you’ve enjoyed reading this and would be interested in getting a highlight of a new open-access paper in your inbox each Sunday, you should consider subscribing:
That’s all for this week, thanks for reading!